ESTABLISHMENT LABS HOLDINGS INC. - Annual Report: 2021 (Form 10-K)
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | ||||
For the fiscal year ended December 31, 2021
or
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | ||||
For the transition period from ___________ to ___________
Commission File Number: 001-38593
Establishment Labs Holdings Inc.
(Exact name of Registrant as specified in its charter)
| British Virgin Islands | Not applicable | |||||||
State or Other Jurisdiction of Incorporation or Organization | I.R.S. Employer Identification No. | |||||||
Building B15 and 25 Coyol Free Zone Alajuela Costa Rica | Not applicable | |||||||
Address of Principal Executive Offices | Zip Code | |||||||
| +506-2434-2400 | ||||||||||||||
| Registrant’s Telephone Number, Including Area Code | ||||||||||||||
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Trading Symbol(s) | Name of Each Exchange on Which Registered | ||||||
| Common Shares, No Par Value | ESTA | The Nasdaq Capital Market | ||||||
Securities registered pursuant to Section 12(g) of the Act:
None.
__________________________________________________
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer”, “smaller reporting company”, and "emerging growth company" in Rule 12b-2 of the Exchange Act. (Check one)
| Large accelerated filer | ☒ | Accelerated filer | ☐ | |||||||||||
| Non-accelerated filer | ☐ | Smaller reporting company | ☐ | |||||||||||
| Emerging growth company | ☐ | |||||||||||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. Yes ☒ No ☐
Indicate by check mark whether the registrant is a shell company (as defined in Exchange Act Rule 12b-2). Yes ☐ No ☒
The aggregate market value of the common stock held by non-affiliates of the registrant on June 30, 2021 was approximately $1,521,927,099. Shares of the registrant’s common stock held by each executive officer, director and holder of 10% or more of the outstanding common stock have been excluded in that such persons may be deemed to be affiliates. This calculation does not reflect a determination that certain persons are affiliates of the registrant for any other purpose. The registrant has no non-voting equity.
As of February 28, 2022, the number of the registrant’s common shares outstanding was 24,124,858.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Registrant’s definitive proxy statement relating to its 2022 annual meeting of shareholders (the “2022 Proxy Statement”) are incorporated by reference into Part III of this Annual Report on Form 10-K where indicated. The 2022 Proxy Statement will be filed with the U.S. Securities and Exchange Commission within 120 days after the end of the fiscal year to which this report relates.
TABLE OF CONTENTS
| Page | ||||||||
i
EXPLANATORY NOTE
In this report, unless the context indicates otherwise, the terms “Establishment Labs,” “Company,” “we”, “us” and “our” refer to Establishment Labs Holdings Inc., a British Virgin Islands entity, and its consolidated subsidiaries.
We own, or have rights to, trademarks and trade names that we use in connection with the operation of our business, including Establishment Labs and our logo as well as other brands such as Motiva Implants, SilkSurface/SmoothSilk, VelvetSurface, ProgressiveGel, TrueMonobloc, BluSeal, Divina, Ergonomix, Ergomonix2, Ergonomix2 Diamond, Motiva MIA and MotivaImagine, among others. Other trademarks and trade names appearing in this report are the property of their respective owners. Solely for your convenience, some of the trademarks and trade names referred to in this report are listed without the ® and TM symbols, but we will assert, to the fullest extent under applicable law, our rights to our trademarks and trade names.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This report contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended or the Exchange Act. You can find many (but not all) of these statements by looking for words such as “approximates,” “believes,” “expects,” “anticipates,” “estimates,” “intends,” “plans,” “would,” “may” or other similar expressions in this report. Any statements that refer to projections of our future financial or operating performance, anticipated trends in our business (including the impact of the COVID-19 outbreak), our goals, strategies, focus and plans, and other characterizations of future events or circumstances, including statements expressing general optimism about future operating results, are forward-looking statements.
We claim the protection of the safe harbor contained in the Private Securities Litigation Reform Act of 1995. We caution investors that any forward-looking statements presented in this report, or that we may make orally or in writing from time to time, are expressions of our beliefs and expectations based on currently available information at the time such statements are made. Such statements are based on assumptions, and the actual outcome will be affected by known and unknown risks, trends, uncertainties and factors that are beyond our control. Although we believe that our assumptions are reasonable, they are not guarantees of future performance. As a result, our actual future results may differ from our expectations, and those differences may be material.
Factors that could cause or contribute to these differences include, among others, those risks and uncertainties discussed below under “Summary Risk Factors” and under Part I, Item 1A. “Risk Factors,” as such risk factors may be amended, updated or superseded from time to time by our subsequent filings with the Securities and Exchange Commission. The risks and uncertainties included herein are not exhaustive, and additional factors could adversely affect our business and financial performance. We operate in a very competitive and rapidly changing environment. New risk factors emerge from time to time, and it is not possible for us to predict all such risk factors, nor can we assess the impact of all such risk factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements.
We are not undertaking any obligation to update any forward-looking statements. Accordingly, investors should use caution in relying on past forward-looking statements, which speak only as of the date they are made.
SUMMARY RISK FACTORS
The following is a summary of certain key risk factors for investors in our securities. You should read this summary together with the more detailed description of risks and uncertainties discussed below under Item 1A. “Risk Factors” before investing in the company.
•The COVID-19 pandemic has adversely affected our business and our financial results, including a material disruption to our operations in fiscal 2020, and may continue to do so for the foreseeable future.
•There is no guarantee that the United States Food and Drug Administration, or FDA, or non-U.S. regulatory agencies will grant approval for our current or future products, and failure to obtain regulatory approvals in the United States and other international jurisdictions, or revocation of approvals in those jurisdictions, will prevent us from marketing our products.
•Even if clinical trials demonstrate acceptable safety and efficacy for Motiva Implants in some patient populations, the FDA or similar regulatory authorities outside the United States may not approve the marketing of Motiva Implants or may approve it with restrictions on the label, which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
1
•In certain large markets, we engage in direct sales efforts. We may fail to maintain and develop our direct sales force, and our revenues and financial outcomes could suffer as a result. Furthermore, our direct sales personnel may not effectively sell our products.
•If we are unable to educate clinicians on the safe, effective and appropriate use of our products and designed surgeries, we may experience increased claims of product liability and may be unable to achieve our expected growth.
•We have a limited operating history in the United States and may face difficulties encountered by companies early in their commercialization in competitive and rapidly evolving markets.
•Our business depends on maintaining our brand and ongoing customer demand for our products and services, and a significant reduction in sentiment or demand could affect our results of operations.
•Any disruption at our existing facilities could adversely affect our business and operating results.
•The medical technology industry is complex and intensely regulated at the federal, state, and local levels and government authorities may determine that we have failed to comply with applicable laws or regulations.
•We rely on a single-source, third-party supplier for medical-grade long-term implantable silicone, which is the primary raw material used in our Motiva Implants. If this supplier were to increase prices for this raw material over time or experience interruptions in its ability to supply us with this raw material, our business, financial condition and results of operations could be adversely affected.
•Negative publicity concerning our products or our competitors’ products, including due to product defects and any resulting litigation, could harm our reputation and reduce demand for silicone breast implants, either of which could adversely impact our financial results and/or share price.
•Recent news coverage has called into question the long-term safety of breast implants and reports of breast implant-associated anaplastic large cell lymphoma (BIA-ALCL) linked to our competitors’ products which have led to regulatory actions regarding macrotextured devices in several countries and the worldwide recall of one of our competitor’s macrotextured implants and tissue expanders. These events may lead to a reduction in the demand for silicone breast implants and could adversely affect our business.
2
PART I
ITEM 1. BUSINESS
Overview
We are a medical technology company focused on improving patient safety and aesthetic outcomes, initially in the breast aesthetics and reconstruction market. We initially incorporated in Costa Rica in 2004 and subsequently reorganized under a parent holding company in the British Virgin Islands in 2013.
Our line of silicone gel-filled breast implants, branded as Motiva Implants, is the centerpiece of our medical technology platform. Our post-market surveillance data (which was not generated in connection with a United States Food and Drug Administration, or FDA, pre-market approval, or PMA, study, but was patient or practitioner reported rather than collected at defined follow-ups) and published third-party data indicates that Motiva Implants show low rates of adverse events (including rupture, capsular contracture, and safety related reoperations) that we believe compare favorably with those of our competitors. We believe the proprietary technologies that differentiate our Motiva Implants enable improved safety and aesthetic outcomes and drive our revenue growth. We have developed other complementary products and services, which are aimed at further enhancing patient outcomes.
Since launching Motiva Implants in October 2010, the majority of our revenue has been generated from sales of our Motiva Implants in cash pay, non-reimbursable, breast augmentation procedures. To date, our Motiva Implants are registered to be sold in more than 80 countries outside of the United States. We currently sell our products via exclusive distributors or our direct sales force (which accounted for approximately 43% of our revenue in 2021) and have introduced five generations of Motiva Implants. We currently commercially sell four product families: (i) Round and Ergonomix Round, (ii) Ergonomix Oval, (iii) Anatomical TrueFixation and (iv) Ergonomix2. Our products incorporate first of-its-kind safety features including: (i) SmoothSilk / SilkSurface (an optimized biocompatible advanced smooth surface that is designed to reduce capsular contracture), (ii) Qid RFID technology (a non-invasive, readable serial number that enables product identification and enhances safety and patient peace of mind), (iii) BluSeal visual barrier layer (a proprietary indicator that allows for verification of complete barrier layer presence) and (iv) TrueMonobloc gel-shell-patch configuration (a highly durable, easy-to-insert performance shell that allows for smaller incisions and smaller scars).
Recent Developments
In April 2021, we completed the enrollment in our one hundred patient Motiva MIA clinical case series in Costa Rica. The Institutional Review Board, or IRB, approved study began in December 2020 subsequent to the initial 2019 case series in Asia. Fifteen board-certified plastic surgeons from Costa Rica, Sweden, England, Brazil, Austria, Italy, Belgium, and the United States participated in the case series. The single-center study is a prospective, single-arm, feasibility study of women 18 years or older in primary minimally invasive breast enhancement.
The Motiva MIA system is designed to provide a minimally invasive breast enhancement procedure in less time and with faster recovery than traditional breast surgery. We have received registration in Costa Rica and a Free Sales Certificate, or FSC, for the components of the Motiva MIA system to begin regulatory approval processes worldwide. The Ergonomix2 Diamond implant used with Motiva MIA obtained CE marking in December 2020 and we have submitted the tools used in the Motiva MIA procedure for CE mark.
In June 2021, we held the groundbreaking ceremony for our new Sulàyöm Innovation Campus in Costa Rica. When complete, the new facility will total approximately 170,000 square feet (16,000 square meters) and will support the company’s continued global growth with additional capacity and capabilities in manufacturing, research and development, or R&D, digital media, training, and medical education. The new campus will be completed in two phases with the first phase expected to cost approximately $35 million. Construction of the new building began following finalization and execution of certain contractual arrangements. The initial phase of the construction of the cold-shell structure is being funded by the Coyol Free Zone, with Establishment Labs having the option to purchase the land and cold shell building. The new facilities will be located in the Coyol Free Zone in Costa Rica and we expect they will add up to 1,000 new jobs over the next several years.
In September 2021, we launched Motiva Flora tissue expander in Europe and other CE mark countries. The Motiva Flora tissue expander offers several notable innovations, including our patented SmoothSilk surface technology, as well as an RFID-enabled, non-magnetic integrated port, both of which offer potential improvements in imaging, treatment, overall clinical outcomes and patient satisfaction.
3
In October 2021, we announced the launch of JOY, a new patient-centric breast aesthetics program. Women who select JOY will receive Establishment Labs’ newest generation Motiva Ergonomix2 implants through a network of highly trained plastic surgeons, as well as, what we believe to be, the most comprehensive patient support program available in the industry. Ergonomix2 incorporates our latest innovations, including our most advanced ultra-high purity chemistries for enhanced device mechanical properties and improved patient ergonomics. Ergonomix2 also features our patented SmoothSilk surface technology, which is the basis of Motiva Implants’ low inflammatory characteristics that have contributed to the lowest capsular contracture rates in the industry. Ergonomix2 has CE mark labeling for use in both aesthetic and reconstruction procedures. JOY includes the Motiva Woman’s Choice Program. This first-of-its-kind program allows women with JOY, subject to certain terms and conditions, to receive financial support from Establishment Labs should they choose to have their implants removed. Women with JOY may also have the option to visit surgeons who commit to reversing the procedure at no additional cost. In addition to the Woman’s Choice Program, JOY also includes Establishment Labs’ Always Confident Warranty, the Motiva 5-Year Extended Warranty, and other benefits.
The Motiva Ergonomix2 implants included with JOY offer all the features of our Ergonomix implants, including SmoothSilk surface technology, ProgressiveGel Ultimate, and RFID enablement, as well as several new technologies, including Motiva SuperSilicones, TrueMonobloc+, and BluSeal+. These advances offer enhanced ergonomy, extra soft feel, and more natural movement. Motiva Ergonomix2 implants are available exclusively through the JOY program.
In February 2019, the FDA granted clearance on the 510(k) submitted for the Motiva Intraoperative Sizers. Sizers are used in breast augmentation or reconstruction procedures to assist in determining the desired breast implant volume and projection before implantation. These devices are part of the platform to support the IDE study and in preparation for the potential PMA approval and commercialization of Motiva Implants in the USA.
In March 2018, we received approval of an investigational device exemption, or IDE, from the FDA to initiate our Motiva Implants clinical trial in the United States for the Motiva Round and Motiva Ergonomix Round product families and the first patient was enrolled in the study in April 2018. In March 2019, we filed our first annual report with the FDA, and our IDE study-defined enrollment targets for the aesthetic cohorts, which include primary augmentation and revision augmentation, had been reached with a total of 450 and 100 subjects, respectively. In August 2019, we announced completion of all surgeries in the aesthetic cohorts. In August 2019, we also announced that we were implementing a bifurcated regulatory strategy in the United States, designed to allow us to initiate rolling submission of data in a PMA from the primary augmentation and revision augmentation cohorts to the FDA, to be supplemented by data from the reconstruction cohorts. We are continuing to enroll subjects in the remaining reconstruction cohorts and plan to complete enrollment of 800 patients in total in the study in fiscal year 2022. In the fourth quarter of 2021, we initiated a modular PMA submission process with the FDA and submitted the first of four expected modules. According to current FDA guidance, a minimum of three years of premarket PMA data must be submitted to support approval of standard silicone gel breast implants; however, the appropriate length of time for collection of premarket study data will be determined by the FDA on a case-by-case basis for each implant after careful consideration of all available clinical and non-clinical data.
Our Market
Breast Augmentation
Breast augmentation surgery remains the leading aesthetic surgical procedure by number of procedures globally. Approximately 1.6 million breast augmentations were performed worldwide in 2020, according to International
4
Society of Aesthetic Plastic Surgery, or ISAPS. The following table lists the top markets by country for total breast augmentations in 2020 according to ISAPS.
| Total Breast Augmentation Procedures | |||||||||||
| Rank * | Country | Procedures | Percentage of World-Wide Total | ||||||||
| 1 | United States | 371,997 | 22.9% | ||||||||
| 2 | Brazil | 172,485 | 10.6% | ||||||||
| 3 | Germany | 67,634 | 4.2% | ||||||||
| 4 | Russia | 59,840 | 3.7% | ||||||||
| 5 | Mexico | 58,312 | 3.6% | ||||||||
| 6 | Argentina | 56,640 | 3.5% | ||||||||
| 7 | Spain | 44,406 | 2.7% | ||||||||
| 8 | Turkey | 39,442 | 2.4% | ||||||||
| 9 | Italy | 39,276 | 2.4% | ||||||||
| 10 | Colombia | 32,724 | 2.0% | ||||||||
* Rankings are based solely on those countries from which a sufficient survey response was received and data was considered to be representative. | |||||||||||
Breast Reconstruction
The American Society of Plastic Surgeons noted in their Plastic Surgery Statistics Report that 137,808 breast reconstructions were performed in 2020. We estimate the 2020 global market was approximately $400 million. The breast reconstruction market is expected to grow and reach approximately $600 million by 2025 at a compound annual growth rate of approximately 7% according to Markets and Markets’ May 2020 Breast Reconstruction Market - Global Forecast to 2025 report due to a combination of increasing incidences of breast cancer and rising awareness of post-mastectomy options available.
Traditional Breast Implants and Their Limitations
Despite the global demand for breast augmentation procedures, there has been relatively little innovation since the 1990s. In 1992, due to emerging safety concerns, the FDA placed a moratorium on sales of silicone breast implants in the United States that was lifted in 2006. This, combined with the ongoing FDA requirement for a PMA on all new breast implants, has discouraged breast implant innovation over the past 30 years. Current products have relatively high adverse event rates, and we believe many do not mimic natural breast tissue. The table below contains key adverse event information from published data from their 10-year prospective clinical trials conducted by the only three companies currently approved to market silicone breast implants in the United States.
Results from primary augmentations
| Sientra 10-Year | Allergan 10-Year | Mentor 10-Year | ||||||||||||||||||
| Number of Patients | N=1,116 Patients | N=455 Patients | N=552 Patients | |||||||||||||||||
Rupture(1) | 8.5% | 9.3% | 24.2% | |||||||||||||||||
| Capsular Contracture | 12.9% | 18.9% | 12.1% | |||||||||||||||||
| Reoperation | 24.0% | 36.1% | 25.5% | |||||||||||||||||
| Kaplan-Meier risk rates were the primary method of analysis for the above data. This table represents the final data from the primary cohort of the same study referenced in the above five- and six-year PMA studies conducted by our competitors. This 10-year data for Sientra, Allergan and Mentor were released in 2018, 2018, and 2015, respectively. | ||||||||||||||||||||
(1) The rupture rates represent the MRI cohort only for each respective study, which consisted of 571 patients for Sientra, 158 patients for Allergan and 202 patients for Mentor. | ||||||||||||||||||||
5
We believe that the improved appearance, feel and patient safety profile of our Motiva Implants provides a strong competitive advantage that will help us to both capture market share and achieve higher patient conversion rates by addressing the key concerns described by patients who choose not to pursue breast augmentation surgery.
Our Competitive Strengths
▪Patient-centric innovative implant technologies. We have developed our Motiva Implants by enhancing and creating novel product components for our implants, and then combining these components into products that deliver improved aesthetic outcomes, increased patient satisfaction and favorable safety profiles.
▪Extensive suite of complementary products and services. Our product portfolio includes innovative products and tools. We believe our designed surgical procedures, such as MotivaHybrid, Motiva MinimalScar and Motiva MIA, will address key unmet needs for both the physician and the patient.
▪Proprietary internal manufacturing processes and capabilities. We manufacture our silicone products in state-of-the-art manufacturing facilities in Costa Rica rather than relying on third-party manufacturers. In these facilities, we utilize our novel 3D imprinted molding method to create proprietary surface features that, in combination with other proprietary materials and methods, differentiate our products from those of our competitors. Our two manufacturing sites have gone through full site inspections and audits under the Medical Device Single Audit Program, or MDSAP, which were carried out by the British Standards Institute, or BSI, an agency which the FDA accepts as a substitute for routine agency inspections. We believe our modern facilities, focus on product quality and deep technological expertise have helped us establish and maintain a brand of consistency, quality and safety.
▪Dynamic worldwide sales platform. We sell our products both through exclusive arrangements with leading local distributors who have strong local surgeon relationships and our direct sales force in key markets such as Brazil and primary markets in Europe. Using this market-specific approach, we have built an effective and efficient worldwide sales platform.
▪Proven management team with expansive industry experience. We have a highly experienced management team that is comprised of leaders from the medical aesthetic market.
Our Growth Strategy
Our goal is to be the global leader in aesthetic surgical implant technology, including breast implants, while improving patient safety through product innovation. The key elements of our strategy include:
▪Expand revenues in existing markets. We believe we can continue to grow market share in our existing markets due to the favorable safety profile and improved aesthetic outcomes of our Motiva Implants.
▪Launch Motiva Implants in additional markets outside the United States. We expect that continued geographic expansion will be a key driver of growth in the near term. In recent years, we started sales through distributors in Australia, Israel, Peru, Russia, Saudi Arabia, Taiwan, Thailand and South Korea, as well as starting direct sales in Brazil, the second largest market for breast augmentations. Expansion into China is expected as early as the second half of 2022.
▪Obtain FDA approval and enter the U.S. market. We are conducting our IDE clinical trial in the United States, with the goal of obtaining approval from the FDA for a premarket application and commercializing our Motiva Implants in the United States. All surgeries have been completed in the aesthetic cohorts. We are continuing to enroll subjects in the remaining primary reconstruction cohort and plan to complete enrollment of a total of 800 patients in the study across 40 sites in the United States, Germany, and Sweden in fiscal 2022.
▪Optimize patient conversion through sales and marketing programs. We employ a multi-faceted marketing strategy that includes social media engagement, conference presence, online advertising and patient and physician education. This approach enables us to engage with and educate patients on the Motiva brand and the benefits of our products, as well as increase clinical efficiency for our physician collaborators. In the future, we expect our social media and online patient and physician education to have important strategic synergies with our designed surgeries, which are promoted globally.
▪Seek out and pursue strategic acquisitions. We intend to seek out other innovative products, services and procedures that satisfy unmet needs in the aesthetics space and complement our existing product portfolio as we believe this can be additive to future revenue growth. We have purchased distributor networks in strategic markets and may acquire other third-party sales organizations in the future. While we have no specific acquisitions or planned licensing agreements currently ongoing, we may engage in
6
these, or other strategic transactions, with the goal of augmenting our existing product portfolio and global footprint.
▪Continue a high level of engagement with key opinion leaders. We promote Motiva Implants, in part, via an extensive and robust calendar of physician education events led by key opinion leaders in the field of aesthetic surgery. In 2021 and 2020, we conducted 206 and 126 events, respectively, through our medical educational platform. We also collaborate actively with respected and influential key opinion leader surgeons to identify and develop new clinical applications for our existing products, as well as new product and strategic opportunities.
7
Our Products and Technologies
The key characteristics of our primary products are described in the table below:
| Product | Motiva Round | Motiva Ergonomix | Motiva Ergonomix2 | Motiva Flora Tissue Expander | ||||||||||
 |  |   |  | |||||||||||
| Description | Round soft silicone-gel filled breast implants | Gravity sensitive round soft silicone-gel-filled breast implants | Gravity sensitive soft silicone-gel-filled breast implants with improved mechanical properties | Breast tissue expander, used to gradually expand a patient’s breast tissue prior to the placement of a long-term breast implant | ||||||||||
| Product Catalog | Available in 160 round catalogs, including four projection heights | Available in 160 round catalogs, including four projection heights | Available in more than 160 round catalogs, including four projection heights; Available in 60 catalogs for Diamond implants | Available in 15 catalogs, with three different heights | ||||||||||
| Key Features | ▪SilkSurface/SmoothSilk shell surface ▪ProgressiveGel PLUS Silicone gel fill ▪TrueMonobloc construction ▪BluSeal shell barrier layer ▪Qid Safety Technology RFID microtransponder | ▪SilkSurface/SmoothSilk shell surface •ProgressiveGel Ultima, Silicone gel fill ▪TrueMonobloc construction ▪BluSeal shell barrier •Qid Safety Technology RFID microtransponder ▪Ergonomy and more natural look | ▪SilkSurface/SmoothSilk shell surface •ProgressiveGel Ultima, Silicone gel fill ▪TrueMonobloc+ construction ▪BluSeal+ shell barrier •Qid Safety Technology RFID microtransponder ▪Motiva SuperSilicones | ▪SilkSurface/SmoothSilk shell surface ▪Anatomical design ▪Compatible with MRI and CT scans ▪Injection site located with RF technology, using the Motiva Port Locator ▪Orientation line observable on the X-Ray ▪Fixation suture tabs | ||||||||||
| Sales Territories | Over 80 countries outside the United States as of December 31, 2021 | |||||||||||||
Motiva Implants
The Motiva breast implants are a class III medical device indicated for breast augmentation and breast reconstruction, including revision surgeries to correct or improve the result of a primary breast augmentation surgery. We launched Motiva Implants commercially in October 2010, and to date we have sold approximately 2.0 million units in various countries outside the United States. Motiva Implants incorporate several proprietary features that we believe contribute to Motiva Implants’ favorable safety profile, natural appearance and feel. Our latest generation of Motiva Implants utilizes our proprietary Gravity Sensitive Ergonomix design, with a round base
8
implant that responds to gravity by shifting its maximum point of projection, offering the more “natural” projection of a shaped implant without the malposition and rotation issues frequently associated with shaped implants. Furthermore, our fill material with the ProgressiveGel platform of silicone gel rheologies consists of highly purified biocompatible gels with specific visco-elastic properties that we believe enables Motiva Implants to respond to the patient’s motion in ways that more closely mimic the appearance, feel and movement of natural breast tissue. Our catalog includes over 1,000 product variations, with round, oval and anatomical shapes, two different surfaces, SmoothSilk and VelvetSurface, and volumes ranging from 95cc to 1060cc, resulting in a wider range of options than those offered by our major competitors.
Ergonomix2 incorporates the latest innovations, including our most advanced ultra-high purity chemistries for enhanced device safety mechanical properties and improved patient ergonomics. Ergonomix2 also features our patented SmoothSilk surface technology, which is the basis of Motiva Implants’ low inflammatory characteristics that have contributed to the lowest capsular contracture rates in the industry. Ergonomix2 was CE marked in December 2020 and labeled for use in both aesthetic and reconstruction procedures.
Shell Surface: SilkSurface/SmoothSilk
The surface topography of the breast implant shell surface varies between commercially available breast implants. Our SmoothSilk surface was designed to improve biocompatibility and to provide for same surface topography around the entire implant for the benefit of the patients. The International Standard Organization, or ISO, through the new April 2018 standard (ISO 14607:2018), created a classification of implant surface textures according to roughness. This standard includes an objective way of defining the difference between smooth, micro and macro surfaces based on roughness average. The topology of SilkSurface/SmoothSilk is characterized under the smooth category, having a low roughness value of approximately 3.09 microns with thousands of contact features per square centimeter, which is significantly lower than the higher limit of the smooth surface clarification defined by ISO.
Our retrospective implant data shows that Motiva Implants have a lower rate of capsular contracture and seromas when compared to available published data from competitors. We believe that these results are due in large part to the proprietary surface of our Motiva Implants. Our proprietary shell surfaces are smoother and have more regular surface features than those of our primary competitors based on several studies using methods such as scanning electron microscopy, profilometry testing and statistical parameters comparisons.
A 2021 published study in Nature Biomedical Engineering led by Professor Robert Langer, Institute Professor at the Massachusetts Institute of Technology (MIT) at David H. Koch Institute for Integrative Cancer Research, indicated that our SmoothSilk can largely suppress the foreign body response and fibrosis provoking the least amount of inflammation in comparison with the other commercially available surfaces. A larger percentage of macrophages in the cell mix indicates an inflammatory response, which is an early step in capsule formation. We believe the more moderate inflammatory response observed on SmoothSilk is responsible for improved biocompatibility and lower complication rates.
In addition, an abstract presented in 2017 by researchers at Montana State University showed less accumulation of both bacteria and biofilm on SmoothSilk in vitro when compared to smoother and textured surfaces. Biofilm formed on implant surfaces increases the risk of bacteria accumulation and capsule formation.
In December 2018, we commissioned an independent report from the French reference laboratory Laboratoire National de Metrologie et d’Essais, or LNE, on the surface characteristics of our Motiva Implants. Based upon its testing, LNE concluded that the SmoothSilk shell surface in the Motiva Implants is considered a smooth surface as defined by ISO 14607:2018 categorization.
9
The graph below shows how the size of our surface features compares with those of our competitors.

ProgressiveGel Family
The proprietary silicone chemistries that comprise our ProgressiveGel family allow for a high degree of cohesiveness and strength but add characteristics such as softness and high ductility that enable movement dynamics more like that of natural breast tissue. We believe that the cohesive properties reduce the likelihood of silicone gel leakage in the event of a rupture in the shell. The strength of the gel is believed to contribute to a reduced frequency of gel fracture, a condition which leads to deformed implant shape and stress on the implant’s shell. While other manufacturers have claimed a “high strength” gel, ours combines a notably high elasticity (the ability to stretch without permanent deformation) with low viscosity, both of which reduce the susceptibility of the implants to rupture while improving their tactile feel and movement dynamics. Additionally, the improved adhesion of the gel to the shell structure avoids the appearance of separation spots, an aesthetic defect commonly seen in competitor products.
In addition to the safety advantages, our ProgressiveGel family, provides for movement characteristics that resemble natural breast tissue. Our later generation Ergonomix products, further mimic natural tissue, with a maximum point of projection that shifts downward to create a natural human breast shape when a patient is standing. This allows our Motiva Implants to provide the more natural aesthetics of “shaped” or “teardrop” implants without the risk of associated drawbacks such as breast deformation from rotation and unnaturally hard tactile feel. The images below illustrate the implants’ ability to change shape depending on the patient’s positioning.
10
 | 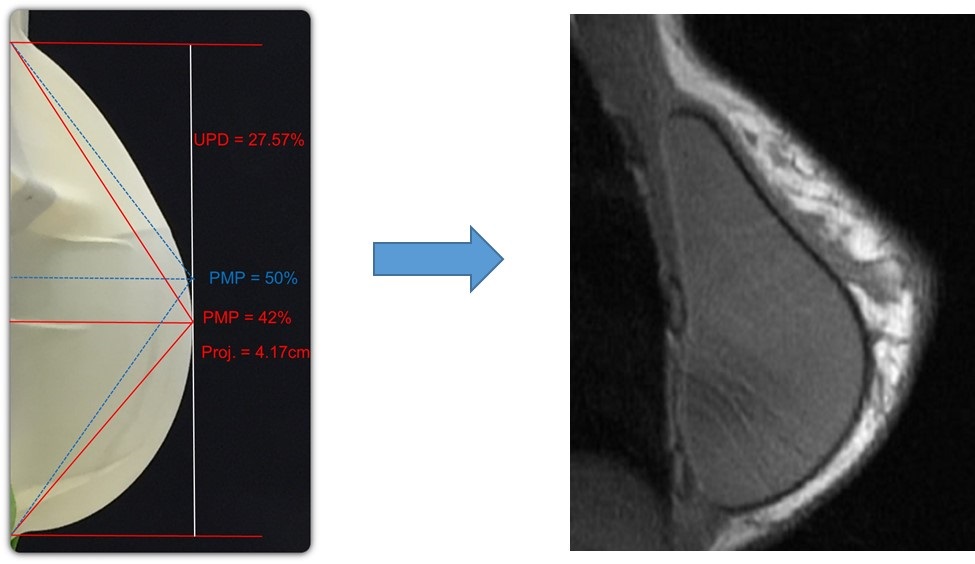 | ||||
TrueMonobloc
Our TrueMonobloc technology, which is incorporated into all generations of Motiva Implants currently sold, combines proprietary chemistry with our proprietary manufacturing techniques to create a shell, gel and other components that are tightly bound to one another. This results in an implant that is more homogeneously elastic and resistant to separation of the gel from the shell, addressing one type of implant failure that can lead to shell ruptures and silicone leaks. This also enables Motiva Implants to be stretched and squeezed to a more significant degree, which we believe currently enables breast augmentation through incision sizes smaller than one inch, compared with the published industry norm of approximately two inches. A surgical technique that we have developed, which we call Motiva Minimally Invasive Augmentation, or Motiva MIA, utilizes our next-generation Ergonomix2 Diamond implant to take advantage of these physical properties to enable a less-invasive procedure for the patient. The implants associated with Motiva MIA received CE Mark approval in December 2020. Instruments and special accessory devices for the Motiva MIA procedure have been developed and are currently awaiting regulatory approval prior to commercialization. The following image shows that TrueMonobloc enables significant manipulation of a Motiva Implant without separation of gel from shell.

RFID Technology
We offer a Radio-Frequency Identification Device microtransponder (also referred to as Qid) that is placed in the filling gel as an optional feature for all implant styles. This microtransponder provides each device with a unique electronic serial number for traceability purposes.
The microtransponder contains only a unique 15-digit code that identifies the product and does not contain any patient information. This microtransponder can be read with a simple pass from our non-invasive and inexpensive reading device, the Qid Safety Technology Reader, and the serial number corresponds with related information in our MotivaImagine database such as implant type, size and other characteristics. Patients can create a secure account, register the products and include applicable patient information either through the MotivaImagine application or our website, to access their implant information. The MotivaImagine application and Motiva Implants website also allow the patient to access the implant warranty information. This traceability is intended to give patients comfort that any future recalls can be positively identified as applying, or not applying, to that patient’s particular implant. This addresses a key concern that often discourages women who are otherwise interested in implants from making the choice to move forward with the surgery. Motiva Implants are currently the
11
only breast implants on the international market with Qid Safety Technology; however, we believe there is an opportunity to sell these microtransponders to other medical device companies in the space.
Each implant’s unique electronic serial number is encoded into the RFID circuitry as part of a three-point authentication system: the microtransponder, the reader and the database. This authentication system prevents unauthorized access to any personal information of the patient and is compliant with FDA regulations.
We also believe that additional functionality can be added to this microtransponder platform. Future potential applications currently under development include temperature sensing as a means of infection detection or pressure sensing as a means of detection of shell rupture.

BluSeal
The Motiva Implant shell is constructed of successive layers of silicone elastomer and a low diffusion barrier layer. The key function of the low diffusion barrier is to prevent diffusion of low molecular weight siloxane species from the implant to the tissues. This barrier layer embeds our BluSeal indicator technology, which is a key feature used during the manufacturing process to verify that the barrier is present in a uniform way around the entire shell. It is also used as a visual quality control and safety measure to minimize potential gel diffusion. This patented manufacturing innovation is intended to highlight any imperfections in the barrier layer coverage with a distinct color. Our BluSeal indicator technology also provides the plastic surgeon with the ability to verify whether the barrier layer has coverage defects or other imperfections before implantation that might lead to post-implantation shell rupture or gel bleed. We believe this is another safety innovation that contributes to our substantially lower reported implant rupture rates as compared to reports for our primary competitors.

Motiva Flora Tissue Expander
The Motiva Flora Tissue Expander is used in breast reconstruction surgery for temporary implantation (less than six months) to gradually expand the breast tissue prior to the placement of a long-term breast implant. After implantation, the device is periodically filled with saline solution via an injection port to increase its volume in order to stretch the skin and create a pocket for breast implant placement. The injection port is dome-shaped and
12
includes an RFID coil, which can be accurately located utilizing the port locator. The Motiva Flora Tissue Expander is the only tissue expander in the market with an integrated RFID port with no magnets, allowing for use of the expander safely alongside MRI scanning. The Motiva Flora received CE mark in June 2020 and has been registered in 33 countries. Motiva Flora also includes the SmoothSilk surface, which provides biocompatibility benefits described above. Our catalog includes 15 variations, including three different heights, and a range of volumes from 260 to 995 cc.
Motiva MIA System for Minimally Invasive Augmentation
We are also developing Motiva MIA — a patient centric procedure designed to allow breast augmentation to be performed under local anesthesia rather than general anesthesia, through smaller incisions, with faster recovery times and a resulting reduction in surgical complications. The Motiva MIA system includes the specially-designed Ergonomix2 Diamond implant, which received CE mark in December 2020, and its proprietary tools, including the Motiva MIA Inflatable Balloon and the Motiva MIA Injector. We received registration in Costa Rica and a Free Sales Certificate, or FSC, for the Motiva MIA devices and we are submitting for regulatory approvals worldwide. Based on third-party commissioned market research, we believe Motiva MIA will be able to attract new customers and expand the market for breast aesthetic procedures.

Puregraft - Autologous Fat Augmentation
Adipose (fat) tissue removed from one area of a patient’s body can be re-injected under the skin of the face, breasts, or in other areas where augmentation and shaping are desired. In the breast augmentation context, there is an unmet need for predictable contouring around the edges of the breast, both with and without volume augmentation via silicone implants. Puregraft LLC’s line of products provides surgeons with a tool for additional contouring around breast implants, which we call MotivaHybrid when used in combination with Motiva Implants and a 3D pre-surgical scan.
MotivaImagine Centers
We utilize our MotivaImagine Center initiative, which are collaborations with plastic surgery clinics whereby we provide them with access to our technologies and the ability to brand themselves as a MotivaImagine Center. In exchange for these services and use of the Motiva branding, each MotivaImagine Center commits to invest in providing prospective patients with a technologically advanced educational experience featuring Motiva Implants and other products in the MotivaImagine product platform.
Designed Surgeries
Our suite of products and technologies enables surgical techniques that we intend to develop and promote as “designed surgeries.” Our first such designed surgery, MotivaHybrid, combines 3D pre-surgical assessment of existing breast tissue volume using a 3D scanning system, together with additional contouring using adipose tissue for more natural balanced results and improved patient satisfaction.
Our second designed surgery, Motiva MinimalScar, allows surgeons to significantly reduce the size of the surgical incision. We have developed Motiva MIA — a family of designed surgeries that enhance breast augmentation in a
13
safer, faster and more predictable way through a small and inconspicuous scar hidden in the axilla. We intend for Motiva MIA to allow breast augmentation procedures to be performed without general anesthesia with faster recovery times and a resulting reduction in surgical complications. In December 2020, we received a CE mark for our Motiva Ergonomix2 Diamond breast implant, which is the implant that will be used in the Motiva MIA procedure. In early 2021, we completed enrollment in our one hundred patient Motiva MIA case series in Costa Rica. The IRB approach study began in December 2020 and one year follow up will be completed in early 2022. Instruments and special accessory devices for the Motiva MIA procedure have been developed and are currently awaiting regulatory approvals prior to commercialization in specific regions. Based on third-party commissioned market research, we believe Motiva MIA will be able to attract new customers and expand the market for breast aesthetic procedures.
Our Clinical Data
11-Year Safety Post-Market Surveillance Data
Dating from the commercial launch of Motiva Implants in October 2010 through December 2021, we have sold approximately 2.0 million breast implants in various countries outside the United States and Canada. We maintain a Quality Management System database to log all complaints received from patients or physicians. From October 2010 through December 2021, a total of 2,094 complaints have been reported, investigated and processed, representing approximately 0.1% of the total Motiva Implants sold through December 2021. There were no reported cases of late seroma, double capsule formation or anaplastic large-cell lymphoma, or ALCL, in this data set, and there were 16 cases of early seroma. The table below shows the rates of rupture, capsular contracture and reoperation for adverse events of our Motiva Implants from the data gathered through December 2021. In contrast to the above competitor data, our data is self-reported rather than collected at mandatory follow-ups and was generated solely for our post-market surveillance instead of in connection with a FDA PMA study. All of these patients were located outside the United States.
| Motiva Implants | ||||||||
| Number of Implants Sold | N= 1,949,663 Implants(1) | |||||||
| Rupture | < 0.1% | |||||||
| Capsular Contracture | < 0.1% | |||||||
| Reoperation for Adverse Events | < 0.1% | |||||||
| Reoperation (All Causes) | N/A(2) | |||||||
| (1) Data is internally tracked on an individual implant basis rather than by patient. | ||||||||
| (2) Complaint database does not capture reoperations for reasons not related to safety. | ||||||||
Independent Clinical Experience
An independent study by Sforza et al., published in the peer-reviewed Aesthetic Surgery Journal in 2017, conducted at a single center, the Hospital Group Ltd.’s Dolan Park Clinic, or Dolan Park, in Bromsgrove, England, between April 2013 and April 2016, reported 5,813 consecutive cases of breast augmentation with Motiva Implants. This independent study was commissioned by Dolan Park’s medical director, Dr. Sforza, who is also a member of our medical advisory board and receives compensation from us in such capacity. The study, conducted by a group of 16 plastic surgeons at Dolan Park, reported overall rates of complication and reoperation of 0.76% over an interval of three years. Beginning in March of 2014, we started supplying our products to Dolan Park under a series of long-term supply agreements with Dolan Park’s affiliated companies. The last supply agreement expired in July of 2019. There were no serious adverse events and no cases of implant rupture for device failure, capsular contracture (Baker III/IV) in primary cases, double capsules, or late seromas. The authors presented consistent real-world data and believe that their free, three-year aftercare system is a strong method for patient retention and follow-up by eliminating any financial limitations for patients to return for follow-up consultations if any issues occur. Anecdotally, the same group of surgeons utilizing the same aftercare system for the last seven years reported substantially different results utilizing other types of silicone breast implants (i.e., non-Motiva Implants). The overall revision rate for this group from 2010 to 2013 utilizing a different, macro-textured, FDA approved implant (N > 10,000) was 8.43%, which is more than 10 times higher than the rate for Motiva Implants reported in this analysis.
14

(1) Names of FDA approved competitors have not been published.
Study to Support a PMA
We are conducting a prospective IDE clinical trial in the United States on our Motiva Round and Motiva Ergonomix Round product families. Our IDE request was approved by the FDA on March 20, 2018 to perform a single open-label, multi-center trial, with follow-up visits available at the time of filing. We will continue to monitor patients for ten years post-implantation. The primary endpoints of the trial will be safety, effectiveness and patient satisfaction. In general, our trial design and patient enrollment are consistent with prior PMA studies conducted by Allergan, Mentor, and Sientra. In August 2019, we announced that we were implementing a bifurcated regulatory strategy in the United States, which is designed to allow us to initiate the rolling submission of data to the FDA from the primary augmentation and revision augmentation cohorts, and then subsequently supplement our PMA with data from the reconstruction cohorts. All the surgeries had been completed in the aesthetics cohort, which include primary augmentation and revision augmentation, with a total of 450 and 100 subjects, respectively. We are continuing to enroll subjects in the remaining reconstruction cohort and plan to enroll a total of 800 patients in the study across 40 sites in the United States, Germany, and Sweden in fiscal year 2022. In the fourth quarter of 2021, we initiated a modular PMA submission process with the FDA and submitted the first of four expected modules.
Sales and Marketing
We primarily derive revenue from sales of our Motiva Implants from two types of customers: (1) medical distributors and (2) direct sales to physicians, hospitals, and clinics. Our products are commercially available in more than 80 countries through exclusive distributors, except in Brazil and several European countries where we sell through our direct sales force. As of December 31, 2021, our sales organization included 117 employees and contractors. All of these sales personnel are supported through a suite of tools, including marketing and training materials, mobile smartphone applications, and access to a robust schedule of physician education events. We also pay significant attention to helping our distributors maintain positive relationships with surgeons and clinics in their respective regions, and to positioning our product in the marketplace as a premium product with consequent premium pricing.
We demonstrate our confidence in Motiva Implants with the Motiva Always Confident Warranty, which offers patients a free replacement for any Motiva Implant that ruptures, for the life of the product. We also replace any implant which is replaced due to capsular contracture of Baker Grade III or IV severity at any time in the first 10 years post-implantation. We also offer an extended warranty at additional cost outside the JOY program, which provides financial assistance of up to $2,500 to cover surgical costs resulting from rupture or capsular contracture.
We employ a multi-faceted marketing strategy that includes social media engagement, conferences, advertisements and education.
15
Intellectual Property
Our success depends at least in part upon our ability to protect our core technology and intellectual property. To accomplish this, we rely on a combination of intellectual property rights, including patents, trade secrets and trademarks, as well as customary contractual protections.
We have assembled a broad portfolio of intellectual property related to our medical device and aesthetics products. We believe this intellectual property, combined with proprietary manufacturing processes and the regulatory approvals we have successfully obtained outside of the United States, provides us with a strong market position. As of December 31, 2021, we own or have rights to 17 issued and 19 pending patents in the United States related to various aspects of our Motiva Implants (such as implant barrier layers, surface texture technology, minimally invasive implant delivery systems, and our Qid Safety Technology radio frequency identification devices). In addition, we own or have rights to 12 issued, two allowed and 85 pending foreign applications and one pending Patent Cooperation Treaty, or PCT, applications. Our owned and licensed patents are expected to expire at various times between February 2025 and February 2039. Our owned and licensed pending applications, if granted, likely would expire between September 2033 and January 2042.
In addition to pursuing patents on our products, we have taken steps to protect our intellectual property and proprietary technology by entering into confidentiality agreements and intellectual property assignment agreements with our employees, consultants, corporate partners, and, when needed, our advisors. Such agreements may not be enforceable or may not provide meaningful protection for our trade secrets or other proprietary information in the event of unauthorized use or disclosure or other breaches of the agreements, and we may not be able to prevent such unauthorized disclosure. Monitoring unauthorized disclosure is difficult, and we do not know whether the steps we have taken to prevent such disclosure are, or will be, adequate. In addition, we intend to expand our international operations, and effective patent, copyright, trademark and trade secret protection may not be available or may be limited in foreign countries.
In general, the medical device industry is characterized by the existence of a large number of patents and frequent allegations and related litigation regarding patent and other intellectual property rights. Third parties, including our competitor companies, may assert patent, copyright, trademark and other intellectual property rights against us, our partners or our customers. Our standard license and other agreements may obligate us to indemnify our partners and customers against such claims. We could incur substantial costs and divert the attention of our management and technical personnel in defending against any such claims. Successful claims of infringement by a third party could prevent us from selling or distributing certain products or performing certain services, require us to expend time and resources to develop non-infringing products, or force us to pay substantial damages, including treble damages if we are found to have willfully infringed patents-royalties or other fees. We cannot assure you that we do not currently infringe, or that we will not in the future infringe, upon any third-party patents or other proprietary rights.
Research and Development
Our goal is to continue to improve our existing products, as well as develop new products and new surgical techniques. We have a highly experienced team and deep customer and key opinion leader relationships. We also have sophisticated internal prototyping and testing equipment. As a result, we have introduced five distinct generations of Motiva Implant product since October 2010, with innovative features added to each successive generation. Further, our efforts included work on both a tissue expander for reconstruction, for which we received a CE Mark in June 2020 and our next generation Ergonomix2 Diamond implant for minimally invasive procedures, for which we received a CE Mark in December 2020.
We have and will continue to work with several institutions in our effort to advance implant technology, and generate additional scientific data to support the improved safety outcomes associated with our products, including:
▪Massachusetts Institute of Technology
▪Medical University of Innsbruck
▪Plastic and Reconstructive Research Center at the University of Manchester
▪Center for Biofilm Engineering of Montana State University
▪The Chair of Plastic Surgery at the School of Medicine and Psychology of Sapienza University of Rome
▪Microscopic Structure Research Center of the University of Costa Rica
16
We have incurred, and expect to continue to incur, significant R&D expenses. Our R&D expenses increased $4.5 million, or 32.8%, to $18.3 million for the year ended December 31, 2021, compared to $13.8 million for the year ended December 31, 2020. Our R&D expenses consist of costs associated with our clinical and post-approval studies, regulatory activity and product development, including the development of Motiva Implants and other current and future aesthetic and reconstruction surgical devices on our product platform.
Implantable RFID Microtransponder Platform
The RFID technology platform that we use in the Qid feature of our Motiva Implants is independently cleared as a system via the FDA’s 510(k) pathway. We are developing more sophisticated functionality using this technology platform. We believe our RFID technology will be an attractive platform for a variety of other applications, including unique device identification for other types of implantable medical devices, functional implantable biosensors, and diagnostic monitoring. Future specific indications include detection of device life cycles (e.g., flexion/contraction cycles for artificial hip and knee joints) and monitoring of analytes such as circulating tumor cells and blood chemistry components. Some of these applications we may choose to develop and commercialize internally, while others may be more appropriately commercialized via partnerships with other medical device companies.
We control all the activities of the development and manufacturing of our Qid Safety Technology RFID transponders. This allows us for adapting to specific needs or new developments in our field.
Manufacturing and Suppliers
Facilities
We manufacture our products in ISO-13485-certified manufacturing facilities located in the Coyol Free Zone office park in Costa Rica, a park populated by a number of international medical device companies and granted tax-advantaged status by the government of Costa Rica. Our newest and largest manufacturing facility opened at the end of 2016 and we began shipping manufactured product from this facility in March 2017. This facility has approximately 28,000 square feet of office space and production areas which are capable of producing over 400,000 implants a year, with state-of-the-art support systems for sustaining production, including an ice-bank system for cooling the controlled air in the clean room and support areas, water-lubricated air compressors for eliminating the presence of oil particulates, heat recovery systems for energy saving, and an energy micro-grid comprised of solar panels and energy-storage batteries. These energy efficient systems generate up to 80% of the total energy consumption of the building, which received LEED Gold Certification by the U.S. Green Building Council in August 2017. Our initial facility was established in 2009 and has about 3,000 square feet of production areas, capable of producing over 100,000 implants a year.
We continue to look for ways to improve manufacturing processes and facility organization to increase capacity in these two current facilities. We completed an internal assessment and identified the potential additional manufacturing capacity of approximately 250,000 implants per year, which we added during fiscal 2021, thereby increasing the efficiencies in our process flow.
In July 2017, both facilities received the MDSAP regulatory certification. MDSAP was established by a coalition of international medical device regulatory authorities including Australia’s TGA, Brazil’s ANVISA, Health Canada, Japan’s MHLW and PMDA and the U.S. FDA. The goal of MDSAP is to allow a single regulatory audit of a medical device manufacturer’s Quality Management System to satisfy the needs of the participating regulatory jurisdictions. This program enables manufacturers to contract with an authorized third-party auditing organization, in our case the British Standards Institute, to conduct a single audit to satisfy the relevant regulatory requirements of the participating regulatory authorities including the FDA, which recognizes MDSAP audit reports as a substitute for FDA Establishment Inspection Reports.
In May 2019, both of our facilities in Coyol Free Zone received the Carbon Neutral certification from the Costa Rican Ministry of Environment, Energy, and Telecommunications, based on the implementation of efficiency-aimed actions such as the reduction of energy consumption through the acquisition of more efficient equipment; the combined use of solar panels, ice banks, and battery storage units; and the avoidance of fossil fuels for our operations.
We are also subject to periodic inspections and audits by various international regulatory and notified bodies, and we believe our past performance in these audits reflects the strength of our quality system and manufacturing controls. We consider this to be a key element of our risk management and business continuity strategies and a competitive advantage as we have full control of the product lifecycle. Our in-house manufacturing team includes
17
over 460 employees, all of whom undergo well defined training programs throughout their period of employment. We believe our manufacturing experience, know-how, and process-related trade secrets are also a competitive advantage.
We are in the process of expanding our manufacturing facilities and corporate offices in the Coyol Free Zone in Costa Rica. The initial $35.3 million project estimate includes approximately 145,000 square feet of facility space and would initially increase our manufacturing capacity by approximately 400,000 units per year, and potentially increase capacity by 800,000 units with an additional incremental $4.6 million investment in manufacturing equipment for a total facility space of approximately 170,000 square feet. We held the groundbreaking ceremony for our new Sulàyöm Innovation Campus in Costa Rica in the second quarter of 2021. Construction on the new building began following finalization and execution of certain contractual arrangements in the third quarter of 2021. The initial phase of construction of the cold-shell structure is being funded by the Coyol Free Zone, with Establishment Labs having the option to purchase the land and cold shell building. See Note 3, “Balance Sheet Accounts” for additional information regarding this construction project and our right to purchase the title to the land and cold shell building currently under construction.
Process
We produce our shell surfaces using a novel 3D negative imprinting molding technique that allows much more precise control over feature size, a uniform distribution of features on the surface, no particles creation, and less unit-to-unit variation. Our primary competitors utilize the “salt-loss” technique or “polyurethane foam imprint” technique. The “salt-loss” technique blows crystals of salt or sugar onto the uncured silicone shell in order to produce surface texture and the “polyurethane foam imprint” technique uses a foreign material to press against the last uncured silicone layer to produce surface features. We believe our 3D negative imprinting technique is more efficient and consistent than the techniques used by our competitors because the application of our advanced smooth surface is integrated with the molding process, rather than requiring a separate, subsequent process.
Suppliers
We source manufacturing inputs from a number of outside suppliers. In particular, we obtain NuSil brand medical-grade silicone from Avantor (previously NuSil Technology LLC), which is a sole-source supplier of such product to the entire silicone breast implant industry. In 2016, we entered into a new supply agreement with NuSil-Avantor, which provides for specified prices per unit of each relevant component; the contract was extended through March 31, 2022 while a new supply agreement is being finalized.
Other critical materials are the silicone patches and other silicone components used for the assembly of our breast implants. All these components are also made with NuSil medical-grade silicone and manufactured by specialized silicone contract manufacturing suppliers. All component suppliers undergo strict quality inspections to ensure these can meet our quality standard. Other important components are the primary packaging polycarbonate trays, the Tyvek sealing lids and packaging. All these components are also critical to maintain integrity of the product throughout its shelf-life and all these suppliers must be qualified and materials must be validated prior to being approved for manufacturing activities. Most suppliers are evaluated annually, and we carry second source supplier activities to ensure business continuity and quality and costs improvement.
Competition
The market for silicone breast implants is relatively concentrated, within Allergan Aesthetics, a division of AbbVie, and Mentor Worldwide LLC, a division of Johnson & Johnson. In the United States, Sientra, Inc. is the only other company with an approved silicone implant product. Internationally, the market is more fragmented, with GC Aesthetics plc, Silimed, Inc., Groupe Sebbin SAS, Hans Biomed Crop., Polytech Health & Aesthetics, and Arion Laboratories.
Our major competitors in the silicone breast implant marketplace are either publicly traded companies or divisions or subsidiaries of publicly traded companies with significantly more market share and resources than we have. These companies have greater financial resources for sales, marketing and product development, broader established relationships with health care providers and third-party payors, and larger and more established distribution networks. In some instances, our competitors also offer products that include features that we do not currently offer in all geographies. Our competitors also have regulatory approval to market and sell their products in countries where we currently do not, notably the United States. In addition, our competitors may offer pricing programs with discounts across their non-breast aesthetic product portfolios.
18
We also face potential future competition from a number of companies, medical researchers and existing medical device companies that may be pursuing new implant technologies. These include non-implant breast augmentation through injections of autologous adipose tissue, new material technologies such as synthetic fillers, and new methods of enhancing and reconstructing the breast.
We believe the primary competitive factors in our current and future markets include:
▪safety and outcomes data generated in clinical studies;
▪regulatory approvals;
▪technological characteristics of products;
▪complementary platforms of non-implant products, such as facial fillers and fat grafting technologies;
▪product price;
▪customer service; and
▪support by key opinion leaders.
Federal Food, Drug, and Cosmetic Act
Breast implants are regulated as Class III medical devices in the United States, and are subject to the Federal Food, Drug, and Cosmetic Act as implemented and enforced by the FDA. The FDA administers requirements covering the design, development, testing, safety, effectiveness, manufacturing, labeling, promotion, advertising, distribution, and postmarket surveillance of medical devices. Medical devices are classified as Class I (lowest risk), II (moderate risk), or III (highest risk). Unless an exemption applies or the product is a Class I device, each medical device that we market must first receive either premarket notification clearance (by filing a 510(k) submission) or premarket approval (by filing a PMA) from the FDA. Breast implants are currently classified as Class III devices requiring an approved PMA for commercial distribution. In addition, certain modifications made to marketed devices also may require 510(k) clearance or approval of a PMA supplement.
The process of obtaining FDA clearance or approval of a medical device can be lengthy and costly. The FDA’s 510(k) clearance process usually takes from three to twelve months, but it can take longer. The process of obtaining PMA approval is much more costly, lengthy, and uncertain, and is generally preceded by the conduct of pre-clinical testing and a well-controlled clinical study. The FDA’s guidance document “Saline, Silicone Gel, and Alternative Breast Implants” currently recommends that a core study, which can be a single, open label, multi-center study, be conducted with ten years or more of prospective patient follow-up. To date, PMAs for silicone breast implants have been submitted for approval to the FDA with a minimum of three years of premarket core study data. Additionally, the FDA will not approve the PMA until it conducts a pre-approval inspection of our manufacturing facility and determines that it is in compliance with good manufacturing practices, as set forth in the FDA’s Quality System Regulation or QSR. The PMA review and approval process generally takes from one to three years but may take longer. The FDA’s guidance document “Saline, Silicone Gel, and Alternative Breast Implants” also states that manufacturers seeking approval of breast implants will be subject to post-approval requirements, which may include, but are not limited to, long-term follow-up of the core clinical study patients, conduct of separate post-approval studies, participation in a patient registry or other studies, and training programs for physicians and surgeons, and periodic reporting requirements.
In addition to regulations governing 510(k) and PMA submissions, we are subject to regulations governing the conduct of clinical investigations, including regulations related to informed consent, Institutional Review Board review and approval, Good Clinical Practices, or GCPs, and labeling of investigational devices. Our clinical study sites are subject to possible inspection by the FDA. We received an IDE approval from the FDA in March 2018, to initiate a clinical trial and our first patient was enrolled in April 2018.
When we initiate commercial distribution of our devices in the United States, we will be subject to FDA device listing and establishment registration, good manufacturing practice requirements as set forth in the QSR, labeling and promotion requirements, reporting of adverse events and device malfunctions, post-approval restrictions or conditions, post-market surveillance requirements, and reporting requirements for product recalls, or corrections or removals in the field. Our manufacturing facilities, as well as those of certain of our suppliers, will be subject to periodic and for-cause inspections by the FDA to verify compliance with the QSR and other regulatory requirements.
19
HIPAA and Other Privacy Laws
We are subject to various laws governing the privacy and security of health information and other personally identifiable information. The Health Insurance Portability and Accountability Act of 1996, or HIPAA, established for the first time comprehensive U.S. federal protection for the privacy and security of protected health information. HIPAA standards apply to “Covered Entities,” which health plans, health care clearing houses, and certain health care providers which conduct certain health care transactions electronically, and to “Business Associates,” entities that perform services on behalf of a Covered Entity that involves the creation, use, maintenance or disclosure of protected health information. Both Covered Entities and Business Associates must have in place administrative, physical, and technical standards to guard against the misuse of protected health information. Some of the institutions and physicians from which we obtain biological specimens that we use in our research and validation work are Covered Entities and must obtain proper authorization from their patients for the subsequent use of those samples and associated clinical information. We may perform future activities that may implicate HIPAA, such as providing clinical laboratory testing services or entering into specific kinds of relationships with a Covered Entity or a Business Associate of a Covered Entity.
Additionally, the Health Information Technology for Economic and Clinical Health Act, enacted as part of the American Recovery and Reinvestment Act of 2009 amended HIPAA by increasing the civil and criminal penalties that may be imposed against Covered Entities, their Business Associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions.
Our activities must also comply with other applicable privacy laws, including the EU General Data Protection Regulation, or GDPR. There are also additional national, state, and provincial privacy laws that impose restrictions on the access, use, and disclosure of personal information, including data that is not protected health information, or are otherwise more stringent than HIPAA. All of these laws may impact our business. If we fail to comply with these privacy laws, or if significant changes in the laws restrict our ability to obtain tissue samples and associated patient information, this could significantly impact our business and our future business plans.
Fraud and Abuse Laws
Antifraud Laws/Overpayments
As participants in national and local health care programs, we may be subject to anti-fraud and abuse laws in various countries. Many of these anti-fraud laws are broad in scope and impose significant penalties for violation. Prohibitions under some of these laws include:
▪the submission of false claims or false information to government programs;
▪deceptive or fraudulent conduct;
▪excessive or unnecessary services, services that do not meet medical necessity, or services at excessive prices; and
▪prohibitions in defrauding private sector health insurers.
Numerous national and local agencies enforce the antifraud and abuse laws. In addition, private insurers may also bring private actions. In some circumstances, private whistleblowers are authorized to bring fraud suits on behalf of the government against providers and are entitled to receive a portion of any final recovery. In addition, we, and our partners, may be subject to foreign laws and regulations and other compliance requirements, including, without limitation, anti-kickback laws, false claims laws and other fraud and abuse laws.
Transparency Laws
We are subject to transparency requirements (also known as “sunshine laws”) in France, including obligations to report payments or transfers of value to, and the nature of the agreements we sign with, a broad class of French healthcare professionals and organizations. If our products are approved in the United States and become eligible for federal government reimbursement, we will also become subject to the Physician Payment Sunshine Act and its amendments and implementing regulations, which require annual reporting of payments and transfers of value to physicians, teaching hospitals and other “covered recipients”, along with physician ownership information. Various states have also implemented regulations prohibiting certain financial interactions with health care professionals or mandating public disclosure of such financial interactions. We may incur significant costs to comply with such laws and regulations now or in the future.
If we or our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil, criminal and administrative penalties, damages, fines, exclusion from participation in government health care programs, additional reporting and
20
government oversight, and the curtailment or restructuring of our operations. As we expand the geographic market for our products, we may be subject to similar national or local laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws, and implementation of corporate compliance programs and reporting of payments or transfers of value to health care professionals. To reduce the risks associated with these various laws and governmental regulations, we have implemented a compliance plan. Although compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, the risks cannot be entirely eliminated. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. Moreover, achieving and sustaining compliance with applicable global privacy, security and fraud laws may prove costly.
International Medical Device Regulations
International marketing of medical devices is subject to foreign government regulations, which vary substantially from country to country.
As of May 2021, medical device products that are marketed in the European Union must comply with the requirements of the Medical Device Regulation, or the MDR. The MDR will essentially operate in the same way as the Medical Device Directive (described below) to ensure a harmonized approach in the European Union to ensuring the safety and performance of medical devices, and failure to comply with the MDR could affect our ability to market and sell our products in the European Union member states. The European Commission is the legislative body responsible for directives, including Regulation (EU) 2017/745 which, once implemented in each member state, must be complied with by manufacturers selling medical products in the EU and the European Economic Area, or EEA. The EU includes most of the major countries in Europe, while other countries, such as Norway, are part of the EEA and European Free Trade Area, or EFTA, respectively, and have voluntarily adopted laws and regulations that mirror those of the EU with respect to medical devices. The EU directives address regulation of the design, manufacture, labeling, clinical studies and post-market vigilance for medical devices. Devices that comply with the requirements of a relevant directive will be entitled to bear the CE conformity marking, indicating that the device conforms to the General Safety and Performance Requirements, or GSPRs, and, accordingly, can be marketed throughout the EU and EEA.
Prior to May 2021, medical device products that were marketed in the European Union were required to comply with the requirements of Medical Device Directive, or the MDD, as implemented in the national legislation of the European Union member states. The MDD, as implemented, provided for a regulatory regime with respect to the design, manufacture, clinical trials, labeling and adverse event reporting for medical devices to ensure that medical devices marketed in the European Union are safe and effective for their intended uses. Medical devices that complied with the MDD, as implemented, are entitled to bear a Conformité Européenne, or CE, marking evidencing such compliance and may be marketed in the European Union. Failure to comply with these domestic and international regulatory requirements could affect our ability to market and sell our products in these countries.
Outside of the EU, regulatory pathways for the marketing of medical devices vary greatly from country to country. In many countries, local regulatory agencies conduct an independent review of medical devices prior to granting marketing approval. For example, in China, approval by the National Medical Products Administration, or NMPA, must be obtained prior to marketing a medical device. In Brazil, the inspections and approvals of products and facilities carried out by the ANVISA and InMetro agencies are required prior to marketing a Class 3a medical device like our Motiva Implants. We received regulatory clearance in Brazil in March 2017 and launched our Motiva Implants commercially in July 2017. The process in such countries may be lengthy and require the expenditure of significant resources, including the conduct of clinical trials. In other countries, the regulatory pathway may be shorter or less costly. The timeline for the introduction of new medical devices is heavily impacted by these various regulations on a country-by-country basis, which may become longer and more costly over time.
Anti-Corruption Laws
We are subject to applicable anti-corruption laws, such as the U.S. Foreign Corrupt Practices Act and the U.K. Bribery Act, and similar anti-corruption laws in the countries in which we distribute our products. Anti-corruption laws generally prohibit offering, promising, giving, or authorizing others to provide anything of value, either directly or indirectly, to a government official or private party in order to influence official action or otherwise gain an unfair business advantage, such as to obtain or retain business. The Foreign Corrupt Practices Act of 1977, as amended, or the FCPA, prohibits any U.S. individual or U.S.-controlled business from paying, offering or
21
authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the United States to comply with accounting provisions requiring us to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations. We have implemented policies, procedures, and internal controls that are designed to comply with these laws and regulations.
Environment
Our manufacturing processes currently require the controlled use of potentially harmful chemicals, including highly flammable solvents and we are subject to inspections and other regulatory requirements, including Costa Rican regulations regarding environmental protection and hazardous and controlled substance controls, among others. Environmental laws and regulations are complex, change frequently and have tended to become more stringent over time. We have incurred, and may continue to incur, significant expenditures to ensure we are in compliance with these laws and regulations. We would be subject to significant penalties for failure to comply with these laws and regulations. For more information, please refer to Section 1A “Risk Factors”.
Human Capital
As of December 31, 2021, we had 746 employees. None of our employees are represented by a labor union or covered by collective bargaining agreements except for employees in Brazil.
The human capital measures and objectives we focus on in managing our business include, as applicable, identifying, recruiting, retaining, incentivizing and integrating our existing and new employees, advisors and consultants. The principal purposes of our equity and cash incentive plans are to attract, retain and reward personnel through the granting of share-based and cash-based compensation awards, in order to increase stockholder value and the success of our company by motivating such individuals to perform to the best of their abilities and achieve our objectives.
We believe that our future success largely depends upon our continued ability to attract and retain highly qualified management and technical personnel. Talent management is critical to our ability to execute on our long-term growth strategy. To facilitate talent attraction and retention, we strive to make our company a safe and rewarding workplace, with opportunities for our employees to grow and develop in their careers, supported by strong compensation and benefits, and by programs that build connections among our employees. We continue to be committed to an inclusive culture which values equity, opportunity, and respect. In support of our inclusive culture, we offer competitive compensation and benefits, including stock awards and strive to recruit a diverse talent pool across all levels of the organization.
ITEM 1A. RISK FACTORS
Investing in our common shares involves a high degree of risk. We operate in a rapidly changing economic and competitive environment that presents numerous risks, many of which are driven by factors that we cannot control or predict. The following risk factors describe circumstances or events that could have a negative effect on our business, financial condition or operating results. You should consider the following risks carefully, together with all the other information in this Annual Report on Form 10-K, including our consolidated financial statements and notes thereto, before you invest in our common shares. If any of the following risks occur, our business, financial condition, or operating results, could be adversely affected. As a result, the trading price of our common shares could decline, and you could lose part or all of your investment. Additional risks and uncertainties not currently known to us or that we currently believe are not material could also impair our business, financial condition or operating results.
Risks Related to COVID-19
The COVID-19 pandemic has adversely affected our business and our financial results, including a material disruption to our operations in fiscal 2020, and may continue to do so for the foreseeable future.
The COVID-19 pandemic has adversely impacted our business, resulting in a material disruption of our operations in fiscal 2020, and we expect the impact to continue through at least the duration of the pandemic as regions respond to local conditions. To date, the impact includes:
•the deferral of procedures using our products;
22
•disruptions or restrictions on the ability of many of our employees and of third parties on which we rely, to work effectively, including “stay-at-home” orders and similar government actions; and
•temporary closures of our facilities and of the facilities of our customers and suppliers.
As jurisdictions throughout the world continue to respond to the pandemic, the degree of the foregoing impacts may increase in scope or magnitude, or we may experience additional adverse effects in one or more regions. Any other outbreaks of contagious diseases or other adverse public health developments in countries where we operate or where our customers or suppliers are located could also have a material and adverse effect on our business, financial condition and results of operations.
Due to the COVID-19 pandemic, surgeons and their patients have been, and in certain regions continue to be, required, or are choosing, to defer elective procedures in which our products otherwise could be used, and many facilities that specialize in the procedures in which our products otherwise could be used have temporarily closed and in some cases continue to be temporarily closed or operating at reduced capacity or hours. In addition, even after the pandemic subsides or governmental orders no longer prohibit or recommend against performing such procedures, patients may continue to defer such procedures due to personal concerns. Further, facilities at which our products typically are used may not reopen or, even if they reopen, patients may elect to have procedures performed at facilities that are, or are perceived to be, lower-risk, such as private surgery centers, and our products may not be approved at such facilities, and we may be unable to have our products approved for use at such facilities on a timely basis, or at all. The effect of the pandemic on the broader economy could also negatively affect demand for elective procedures using our products, both in the near- and long-term.
Workforce limitations and travel restrictions resulting from government actions taken to contain the spread of COVID-19 have and will continue to adversely affect almost every aspect of our business. If a significant percentage of our workforce, or of the workforce of third parties on which we rely, cannot work, including because of illness or travel or government restrictions, our operations will be negatively affected. Because of government restrictions and social distancing guidelines in many countries around the world, there is an increased reliance on working from home for our workforce and on the workforce of third parties on which we rely. For example, most of our sales personnel and third-party agents currently are working largely using virtual and online engagement tools and tactics, which may be less effective than our typical in-person sales and marketing programs. In addition, we reduced access to our hands-on surgeon trainings, which, in turn, adversely impacted our ability to educate and train surgeons on the proper use of our products, which may make surgeons less comfortable using, and therefore less likely to use, our products. We expect that governmental mandates or other restrictions will also limit our ability to develop, and therefore launch, the products we believe will drive our future revenue growth on the timelines we anticipated previously and could also delay the planned launch of products in 2022 and beyond. It may also cause us not to submit required filings on our previous timetables, including with the FDA, or other regulatory bodies, both in the U.S. and outside the U.S. The continued spread of COVID-19 has adversely impacted our IDE clinical trial operations in the United States, including our ability to recruit and retain patients and principal investigators and site staff who, as healthcare providers, have heightened exposure to COVID-19. In addition, changes impacting workforce function at the FDA and other regulatory bodies, as well as changes impacting workforce function at the facilities at which we seek to have new products approved for use, could adversely impact the timing of when our new products are cleared for marketing and approved for use, either of which would adversely impact the timing of our ability to sell these new products and would have a material and adverse effect on our revenue growth.
Further, disruptions in the manufacture and distribution of our products or in our supply chain may occur as a result of the COVID-19 pandemic, including for the reasons above, or other events that result in staffing shortages, production slowdowns, stoppages, or disruptions in delivery systems, any of which could materially and adversely affect our ability to manufacture or distribute our products, or to obtain the raw materials and supplies necessary to manufacture and distribute our products, in a timely manner, or at all.
We may also experience other unknown adverse impacts from the COVID-19 pandemic that cannot be predicted. For example, hospitals and other facilities at which we sell our products may renegotiate their purchase prices, including as a result of, or the perception that they may be suffering, financial difficulty as a result of the pandemic. Similarly, facilities at which we seek to sell our products in the future may require price reductions relative to the price at which we previously expected to sell our products. Reduction in the prices at which we sell products to existing customers may have a material and adverse effect on our future financial results and reductions in the
23
prices at which we expected to sell products would have a material and adverse effect on our expectations for revenue growth.
Further, the global capital markets experienced, and we expect will continue to experience, disruption and volatility due to the COVID-19 pandemic, adversely impacting access to capital not only for us, but also for our customers and suppliers who need access to capital. Their inability to access capital in a timely manner, or at all, could adversely impact demand for our products and/or adversely impact our ability to manufacture or supply our products, any of which could have a material and adverse effect on our business.
The full extent to which the COVID-19 pandemic will, directly or indirectly, impact our business, results of operations and financial condition, including our sales, expenses, supply chain integrity, manufacturing capability, research and development activities, and employee-related compensation, is currently highly uncertain and cannot be predicted with reasonable accuracy at this time and will depend on future developments that are also highly uncertain and cannot be predicted with reasonable accuracy at this time, including, without limitation:
•new information that may emerge concerning COVID-19, its contagiousness or virulence;
•resurgences in COVID-19 transmission and infection following the easing or lifting of governmental or other restrictions or following resumption of surgical procedures, whether as a result thereof, as a result of reinfection, as a result of a delay in the emergence of symptoms following infection (or reinfection) by COVID-19, or as a result of COVID-19’s ability to lay dormant following infection (or reinfection), and the adverse impact the foregoing may have on our business and financial condition, including because of the adverse impact on patients’ willingness to undergo procedures in which our products could be used;
•actions required or recommended to contain or treat COVID-19, in light of any or all of the foregoing or other as-yet unanticipated developments, whether related to COVID-19 directly or indirectly; and
•the direct and indirect economic impact, both domestically and abroad, of the COVID-19 pandemic as a result of any or all of the foregoing, including actions taken by local, state, national and international governmental agencies, whether such impact affects customers, suppliers, or markets generally.
Risks Related to the Development and Commercialization of Our Products
We have a limited operating history in the United States and may face difficulties encountered by companies early in their commercialization in competitive and rapidly evolving markets.
Our Motiva Implants have been marketed solely in countries outside of the United States since October 2010, and as such, we have a limited operating history upon which to evaluate our business and forecast our future net sales and operating results. In assessing our business prospects, you should consider the various risks and difficulties frequently encountered by companies early in their commercialization in competitive markets, particularly companies that develop and sell medical devices. These risks include our ability to:
•implement and execute our business strategy;
•expand and improve the productivity of our direct sales force, distributors and marketing programs to grow sales of our products;
•increase awareness of our brands and build loyalty among plastic surgeons and patients;
•manage expanding operations;
•respond effectively to competitive pressures and developments;
•enhance our existing products and develop new products;
•maintain and obtain regulatory clearance or approval of our existing products and commercialize new products;
•respond to changing regulations associated with medical devices across all geographies;
•perform clinical trials with respect to our existing products and any new products;
•attract, retain and motivate qualified personnel in various areas of our business; and
•obtain and maintain coverage and adequate levels of reimbursement for our products.
24
Due to our limited operating history in the United States, we may not have the institutional knowledge or experience to be able to effectively address these and other risks that we may face. In addition, we may not be able to develop insights into trends that could emerge and negatively affect our business and may fail to respond effectively to those trends. As a result of these or other risks, we may not be able to execute key components of our business strategy, and our business, financial condition and operating results may suffer.
Our success depends, in part, on our ability to continue to enhance our existing products and services and develop or commercialize new products and services that respond to customer needs and preferences, which we expect will require us to incur significant expenses.
In recent years, we have incurred significant costs in connection with the development of Motiva Implants, the Motiva MIA technology, and other products and services. We expect our research and development expenses to increase significantly as we continue with our IDE clinical trial in the United States. We will also incur significant expenses to expand our sales and marketing organization to support sales of Motiva Implants, including but not limited to a direct sales force in Brazil and several European countries, as well as other products outside the United States and Canada.
We may not be able to compete effectively with our competitors, and ultimately satisfy the needs and preferences of our customers, unless we can continue to enhance existing products and develop or acquire new innovative products and services. Product development requires the investment of significant financial, technological and other resources. Product improvements and new product introductions also require significant planning, design, development and testing at the product and manufacturing process levels. We may not be able to timely or effectively develop product improvements or new products and services. Likewise, we may not be able to acquire new products on terms that are acceptable to us, or at all. Furthermore, in most countries, we need to obtain regulatory approval in order to market and sell our products, which may limit our ability to act quickly in scaling commercialization in those countries, including the United States. Our competitors’ new products may beat our products to market, be more effective or safer or have new features, obtain better market acceptance or render our products and services obsolete. Any new or modified products and services that we develop may not receive regulatory clearance or approval, or achieve market acceptance or otherwise generate any meaningful sales or profits for us.
Motiva Implants are not currently approved for commercial sale in the United States. Obtaining such approval is costly and time consuming, and we may not obtain the regulatory approval required to sell our products in the United States.
Neither we, nor any future collaboration partner, can commercialize Motiva Implants in the United States without first obtaining regulatory approval for the product from the FDA. In the EU and other countries, we previously obtained a CE Mark, before making Motiva Implants available for commercial sale. FDA guidance on silicone breast implants mandates approval via the PMA process. Extensive preclinical and clinical testing will be required to support the PMA. At least one well-controlled clinical trial is required for approval, such as the one we began in April 2018, which will require us to commit significant financial and personnel resources. Additionally, we will be required to commit to significant and costly post-approval requirements, which will include follow-up of our clinical trial patients for up to ten years, creation of a patient registry, and/or other studies, and implementation of training programs for physicians. We may be unable to fund, enroll, or complete such trials in a timely fashion, or at all, and we may have an insufficient number of enrolled patients follow up as instructed. The results of clinical studies may not be favorable enough to support marketing approval in the United States, or may raise other questions (pertaining, for example, to product safety or effectiveness) that jeopardizes our current approvals for sale in other territories. The FDA approval process will take at least several years to complete, and FDA approval may never be obtained. We must also demonstrate that our manufacturing facilities, processes and controls are adequate to support FDA approval and that our clinical investigators complied with good clinical practices in the conduct of the clinical trial for our Motiva Implants.
Furthermore, FDA regulatory approval is not a guarantee, and the filing and approval process itself is expensive and may take several years. The FDA also has substantial discretion in the approval process. Despite the time and expense exerted, failure may occur at any stage, and we could encounter problems that cause us to abandon or repeat clinical studies, including our ongoing IDE clinical trial that commenced in April 2018. The FDA can delay, limit, or deny approval of a product candidate for many reasons, including, but not limited, to:
•a product candidate may not be deemed to be safe and effective;
25
•FDA officials may not find the data from clinical and preclinical studies sufficient;
•the FDA may not approve our or our suppliers’ processes or facilities;
•the FDA may consider clinical studies inadequate and the data inadequate if, among other things, appropriate steps have not been taken in the design, conduct, reporting and analysis of the studies to minimize bias; or
•the FDA may change its approval policies or adopt new regulations.
If Motiva Implants, or our future products, fail to demonstrate safety and efficacy in further clinical studies that may be required for U.S. approval, or do not gain regulatory approval, our business and results of operations will be harmed.
Moreover, obtaining regulatory approval for marketing of our products in one country does not ensure we will be able to obtain regulatory approval in other countries, while a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in other countries.
There is no guarantee that the United States Food and Drug Administration, or FDA, or non-U.S. regulatory agencies will grant approval for our current or future products, and failure to obtain regulatory approvals in the United States and other international jurisdictions, or revocation of approvals in those jurisdictions, will prevent us from marketing our products.
We intend to seek additional distribution and marketing partners for Motiva Implants and may market specific products only in international markets. We have obtained a CE Mark for Motiva Implants and are therefore authorized to sell in the EU; however, in order to market in regions such as the Asia Pacific region and many other jurisdictions, we must obtain separate regulatory approvals. The approval procedures vary among countries and can involve additional clinical testing, and the time required to obtain approvals may differ from that required to obtain the CE Mark or FDA approval. Moreover, clinical studies or manufacturing processes conducted in one country may not be accepted by regulatory authorities in other countries. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one or more international regulatory authorities does not ensure approval by regulatory authorities in other countries or by the FDA. However, a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. An international regulatory approval process may include all of the risks associated with obtaining FDA approval. We may not obtain international regulatory approvals on a timely basis, if at all. We may not be able to file for regulatory approvals and, even if we file, we may not receive necessary approvals to commercialize our products in any market.
Before obtaining regulatory approval for the sale of a planned product, we may be required to conduct extensive preclinical and clinical studies to demonstrate the safety and efficacy of our planned products in human patients. Clinical studies can be expensive, difficult to design and implement, can take many years to complete, and are uncertain as to outcome. A failure of one or more of our clinical studies could occur at any stage of testing. In connection with the initiation of a clinical study in the United States, we filed an IDE application in 2017, which was approved in March 2018 and our first patient was enrolled in April 2018. Our ongoing U.S. IDE trial may take longer to enroll than anticipated, may be stopped for unforeseen safety issues or may not be successful in meeting its endpoints, in which case our U.S. regulatory pathway would require subsequent additional clinical trials.
Numerous unforeseen events during, or as a result of, preclinical and clinical studies could occur, which would delay or prevent our ability to receive regulatory approval or commercialize Motiva Implants or any of our planned products, including the following:
•clinical studies may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical studies or abandon product development programs;
•the number of patients required for clinical studies may be larger than we anticipate, enrollment in these clinical studies may be insufficient or slower than we anticipate, or patients may drop out of these clinical studies at a higher rate than we anticipate;
•the cost of clinical studies may be greater than we anticipate;
•third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all;
26
•we might suspend or terminate clinical studies of our planned products for various reasons, including a finding that our planned products have unanticipated serious side effects or other unexpected characteristics, or that the study subjects are being exposed to unacceptable health risks;
•regulators may not approve our proposed clinical development plans;
•regulators or independent institutional review boards, or IRBs, may not authorize us or our investigators to commence a clinical study or conduct a clinical study at a prospective study site;
•regulators or IRBs may require that we, or our investigators, suspend or terminate clinical studies for various reasons, including noncompliance with regulatory requirements;
•regulators may determine that the clinical data submitted to support our request for approval is unreliable or incomplete as a result of any number of factors, including potential financial bias associated with equity holdings in the Company by study investigators, or significant payments by the Company to study investigators for consulting work, which may result in regulators requesting further data analysis or other confirmatory studies to be performed, or determining the data does not support regulatory approval;
•regulators in countries where Motiva Implants are currently marketed may require that we suspend commercial distribution if there is noncompliance with regulatory requirements or safety concerns;
•regulators in countries where Motiva Implants are currently marketed may suspend commercial distribution of silicone breast implants due to safety or other concerns generally applicable to the product category;
•the supply or quality of our planned products or other materials necessary to conduct clinical studies of our planned products may be insufficient or inadequate; and/or
•the enactment of new regulatory requirements in Europe under the new Medical Device Regulation may make approval times longer and standards more difficult to pass.
If we, or any future collaboration partner, are required to conduct additional clinical trials or other testing of Motiva Implants or any planned products, those clinical studies or other testing may not be successfully completed. Additionally, if the results of these studies or tests are not positive, or if they raise safety concerns, we may:
•be delayed in obtaining marketing approvals for Motiva Implants or our planned products;
•not obtain marketing approval at all;
•obtain approval for indications that are not as broad as intended;
•have a product removed from the market after obtaining marketing approval;
•be subject to additional post-marketing testing requirements; and/or
•be subject to restrictions on how the product is distributed or used.
Even if we obtain regulatory approvals or clearances in a jurisdiction, our products may be removed from the market due to a variety of factors, including adverse events, recalls, suspension of regulatory clearance to sell, or other factors. For example, during the summer of 2016 while we were transitioning from one notified body to another, our CE Mark for Motiva Implants was temporarily not in force. We expect that the initial U.S. approval will be subject to a lengthy and expensive follow-up period, during which we must monitor patients enrolled in clinical studies and collect data on their safety outcomes. Even if FDA approval is obtained, FDA has authority to impose postmarket approval conditions, which can include (i) restrictions on device’s sale, distribution, or use, (ii) continuing evaluation of the device’s safety and efficacy, (iii) additional warning/hazard labeling requirements, (iv) significant record management, (v) periodic reporting requirements, and (vi) any other requirements the FDA determines necessary to provide reasonable assurance of the device’s safety and effectiveness. Completion of this follow-up study, in a manner which results in data sufficient to maintain FDA approval, is subject to multiple risks, many of which are outside of our control. These include, but are not limited to, our ability to fund the ongoing study from our operations or via additional fundraising; study participants’ willingness and ability to return for follow-up study visits; and maintenance of a suitable study database over a long period of time. Even if completed and appropriately evaluated, the study follow-up may reveal safety or other issues that impact the approved labeling or may result in withdrawal of Motiva Implants from the marketplace in the United States or elsewhere.
27
Although we launched Motiva Implants commercially in October 2010 and have sold approximately 2.0 million units to date in various countries outside the United States, we do not have as much post-market surveillance data as our competitors and may not have clearly identified all possible or actual risks of our products. Furthermore, if our clinical trials do not produce patient data that compares favorably with breast implants that are already on the market, physicians and patients may opt to not use our products, and our business would suffer.
Our product development costs will also increase if we experience delays to our clinical trials or approvals. We do not know whether any clinical studies will begin as planned, will need to be restructured, or will be completed on schedule, or at all.
Significant clinical study delays could allow our competitors to bring products to market before we do, which would impair our ability to commercialize our planned products and harm our business and results of operations.
Even if clinical trials demonstrate acceptable safety and efficacy for Motiva Implants in some patient populations, the FDA or similar regulatory authorities outside the United States may not approve the marketing of Motiva Implants or may approve it with restrictions on the label, which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
It is possible the FDA or similar regulatory authorities may not consider the results of our clinical trials to be sufficient for approval of Motiva Implants for our desired indications for use. Guidance issued by the FDA in 2006 suggests that a single well-controlled study is required for approval of a new silicone breast implant. The FDA may nonetheless require that we conduct additional clinical studies, possibly using a different clinical study design.
Moreover, even if the FDA or other regulatory authorities approve the marketing of Motiva Implants, the approval may include additional restrictions on the label that could make Motiva Implants less attractive to physicians and patients compared to other products that may be approved for broader indications, which could limit potential sales of Motiva Implants.
If we fail to obtain FDA or other regulatory approval of Motiva Implants, or if the approval is narrower than what we seek, it could impair our ability to realize value from Motiva Implants, and therefore may have a material adverse effect on our business, financial condition, results of operations and growth prospects.
Any future distribution or commercialization agreements we may enter into with respect to our current or planned products may place the development of these products outside our control, or may otherwise be on terms unfavorable to us.
We may enter into additional distribution or commercialization agreements with third parties with respect to our current or planned products, for commercialization in or outside the United States. Our likely collaborators for any distribution, marketing, licensing or other collaboration arrangements include large and mid-size medical device and diagnostic companies, regional and national medical device and diagnostic companies, and distribution or group purchasing organizations. We will have limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of our planned products. Our ability to generate revenue from these arrangements will depend in part on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements.
Collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable planned products. Collaborators may own or co-own intellectual property covering our products that results from our collaboration with them. In such cases, we would not have the exclusive right to commercialize such intellectual property.
Any termination or disruption of collaborations could result in delays in the development of planned products, increases in our costs to develop the planned products or the termination of development of a planned product.
If we are unable to educate clinicians on the safe, effective and appropriate use of our products and designed surgeries, we may experience increased claims of product liability and may be unable to achieve our expected growth.
We make extensive physician medical education resources available to clinicians in an effort to ensure that they have access to current treatment methodologies, are aware of the advantages and risks of our Motiva Implants and other products, and are educated regarding the safe and appropriate use of our products. It is critical to the success of our business to broadly educate clinicians who use or desire to use our products to provide them with adequate instructions in the appropriate use of our products and designed surgeries. Certain of our products
28
require the use of specialized techniques which may not be covered in medical school curricula and/or product-specific knowledge. For example, metal implant such as screws or artificial joints, produce an artifact when magnetic resonance imaging, or MRI, is used to image the area in which the object resides. Our Qid Safety Technology microtransponder embedded in certain Motiva Implants contains metal and causes an artifact that can affect breast cancer screening using MRI, and this artifact is not present in other imaging modalities such as breast ultrasound and film or digital mammography. It is important that we educate physicians and patients on the risks associated with MRI artifacts and how to mitigate them if they choose to utilize Motiva Implants that contain a Qid microtransponder. Failure to provide adequate training and education could result in, among other things, unsatisfactory patient outcomes, patient injury, negative publicity or increased product liability claims or lawsuits against the company, any of which could have a material and adverse effect on our business and reputation. Claims against the company may occur even if such claims are without merit and/or no product defect is present, due to improper surgical technique, inappropriate use of our products, or other lack of awareness regarding the safe and effective use of our products. If we fail to educate physicians and patients about any of these factors, they may make decisions or conclusions regarding Motiva Implants without full knowledge of the risks and benefits or may view our Motiva Implants negatively.
As part of our effort to educate and train plastic surgeons through our medical educational platform, we completed 206 and 126 medical training sessions worldwide during 2021 and 2020, respectively. If we are unable to offer, or if we experience a delay in offering, medical training sessions, we may experience reduced or slower than expected adoption of our products. Although since the outbreak of the global COVID-19 pandemic we have offered virtual training sessions through our medical educational platform, the limited ability to provide in-person programs to surgeons may reduce the effectiveness of, and interest in, our medical education efforts.
Commercial success of Motiva Implants in the United States or elsewhere depends on our ability to accurately forecast customer demand and manufacture sufficient quantities of product in the implant sizes that patients and physicians request, and to manage inventory effectively and the failure to do so could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
Manufacturing of silicone breast implants requires costly capital equipment and a highly skilled workforce. There is a significant lead time to build and certify a new manufacturing facility. Until 2017, we had one manufacturing facility in Costa Rica, and we experienced inventory shortages from time-to-time that impaired our ability to meet market demand. In March 2017, our second manufacturing facility, also located in Costa Rica, became operational, and we received certification under the multi-country MDSAP protocol and began shipping saleable product. Although we believe our new, larger manufacturing facility, in combination with our first facility, will give us adequate manufacturing capacity to meet demand for at least the next two years, we have, in the past, been unable to fill all incoming orders to meet growing demand. We began construction on the expansion of one of our facilities in Costa Rica during the third quarter of 2021. If this expansion is not completed in a timely manner, our ability to fill incoming orders may be adversely impacted. In addition, if we obtain FDA approval, we will likely need to obtain additional manufacturing capacity prior to any commercialization of our Motiva Implants in the United States. If demand increases faster than we expect, or if we are unable to produce the quantity of goods that we expect with our current facilities, we may not be able to grow revenue at an optimal rate. There may be other negative effects from supply shortages, including loss of our reputation in the marketplace and a negative impact on our relationships with our distributors.
On the other hand, if demand for our products declines, or if market supply surpasses demand, we may not be able to reduce manufacturing expenses or overhead costs proportionately. We have invested significantly in our manufacturing capacity in order to vertically integrate our business. If an increase in supply outpaces the increase in market demand, or if demand decreases, the resulting oversupply could adversely impact our sales and result in the underutilization of our manufacturing capacity, higher inventory carrying costs and associated working capital, changes in revenue mix, and/or price erosion, any of which would lower our margins and adversely impact our financial results.
Risks Related to Our Business, Industry and Operations
We expect to incur losses for the foreseeable future, and our ability to achieve and maintain profitability depends on the commercial success of our Motiva Implants, which accounted for approximately 98% of
29
our revenues for each of the years ended December 31, 2021 and 2020 and we expect our revenues to continue to be driven primarily by sales of these products.
We have incurred losses to date and expect to continue to incur losses for the foreseeable future. Sales of our Motiva Implants accounted for approximately 98% of our revenues for each of the years ended December 31, 2021 and December 31, 2020 and we expect our revenues to continue to be driven primarily by sales of these products. In order to achieve and sustain profitability, our revenues from these products will need to grow beyond the levels we have achieved in the past. If physicians and/or patients do not perceive our products to be competitive in features and safety when compared to other products in the market, or if demand for our Motiva Implants or for breast implants in general decreases, we may fail to achieve sales levels that provide for future profitability.
Our ability to successfully market Motiva Implants and our other current and future offerings depends on numerous factors, including but not limited to:
•the outcomes of current and future clinical studies of Motiva Implants, including our ongoing IDE clinical trial, to demonstrate our products’ value in improving safety outcomes and/or patient satisfaction;
•acceptance of Motiva Implants as safe and effective by patients, caregivers and the medical community;
•an acceptable safety profile of Motiva Implants in the global market;
•whether key thought leaders in the medical community accept that such clinical studies are sufficiently meaningful to influence their or their patients’ choices of product;
•maintenance of our existing regulatory approvals and expansion of the geographies in which we have regulatory approvals;
•designing commercially viable processes at a scale sufficient to meet anticipated demand at an adequate cost of manufacturing, and that are compliant with ISO 13485 Quality Management System requirements and/or good manufacturing practice, or GMP, requirements, as set forth in the FDA’s Quality System Regulation, Brazilian and other international regulations;
•our success in educating physicians and patients about the benefits, administration and use of Motiva Implants;
•the availability, perceived advantages, relative cost, relative safety and relative efficacy of alternative and competing treatments;
•the willingness of patients to pay out-of-pocket for breast augmentation and reconstruction procedures in the absence of coverage and reimbursement for such procedures;
•the success of our internal sales and marketing organization and the sales forces of our distributors; and
•continued demand for breast augmentation and reconstruction procedures using silicone implants, which may be adversely affected by events involving either our products or those of our competitors, including FDA warnings to patients regarding Breast Implant-Associated Anaplastic Large Cell Lymphoma, or BIA-ALCL.
Some of these factors are beyond our control. If we are unable to continue to commercialize Motiva Implants and our other products, or unable to obtain a partner to commercialize them, we may not be able to produce any incremental revenues related to Motiva Implants and our other products. This would result in an adverse effect on our business, financial condition, results of operations and growth prospects.
We have incurred net operating losses in the past and expect to incur net operating losses for the foreseeable future.
We have incurred net operating losses since our inception, and we continue to incur significant research and development and general and administrative expenses related to our operations. We do not expect to be profitable in 2022, and in future years we expect to incur significant research and development expenses related to, among other things, the IDE clinical study of Motiva Implants in the United States. Investment in medical device product development, particularly clinical studies, is highly speculative. It entails substantial upfront capital expenditures and significant risk that any potential planned product will fail to demonstrate adequate accuracy or
30
clinical utility. We may not be profitable for some time. As of December 31, 2021, we had an accumulated deficit of $206.4 million.
We expect that our future financial results will depend primarily on our success in launching, selling and supporting Motiva Implants and other products that are part of our product platform. This will require us to be successful in a range of activities, including manufacturing, marketing, and selling Motiva Implants. We may not succeed in these activities and may never generate revenue that is sufficient to be profitable in the future. Even if we are profitable, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to achieve sustained profitability would depress the value of our company and could impair our ability to raise capital, expand our business, diversify our planned products, market our current and planned products, or continue our operations.
We may need additional funds to support our operations, and such funding may not be available to us on acceptable terms, or at all, which would force us to delay, reduce or suspend our planned development and commercialization efforts. Raising additional capital may subject us to unfavorable terms, cause dilution to our existing shareholders, restrict our operations, or require us to relinquish rights to our products and technologies.
Our operations have consumed substantial amounts of cash since our inception, and we expect to incur significant expenses in connection with our planned research, development and product commercialization efforts. We believe that our available cash and cash from operations will be sufficient to satisfy our liquidity requirements for at least the next 12 months and beyond. If our available cash resources, net proceeds from our follow-on public offering and anticipated cash flow from operations are insufficient to satisfy our liquidity requirements, we may seek to sell equity or convertible debt securities, enter into a credit facility or another form of third-party funding, or seek other debt financing. However, we are subject to restrictive covenants under the Madryn Credit Agreement which restrict our ability to incur additional debt. Any failure to raise the funds necessary to support our operations may force us to delay, reduce or suspend our planned clinical trials, research and development programs, or other commercialization efforts.
To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership may be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a shareholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take certain actions, such as incurring additional debt, making capital expenditures or declaring dividends.
If we raise additional funds through collaborations, strategic collaborations or partnership, or marketing, distribution or licensing arrangements with third parties, we may be required to do so at an earlier stage than would otherwise be ideal and/or may have to limit valuable rights to our intellectual property, technologies, products, or future revenue streams, or grant licenses or other rights on terms that are not favorable to us. Furthermore, any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our products.
Our business depends on maintaining our brand and ongoing customer demand for our products and services, and a significant reduction in sentiment or demand could affect our results of operations.
Our success depends on the reputation of our brands, which depends on factors such as the safety and quality of our products, our communication activities, including marketing and education efforts, and our management of our customer experience. Maintaining, promoting and positioning our brands is important to expanding our customer base. This will depend largely on the success of our education and marketing efforts and our ability to provide a consistent, high-quality customer experience.
We may need to make substantial investments in the areas of education and marketing in order to maintain and enhance our brands. Ineffective marketing, negative publicity, significant discounts by our competitors, product defects and related liability litigation, failure to obtain regulatory clearance for our products, counterfeit products, unfair labor practices and failure to protect the intellectual property rights in our brands are some of the potential threats to the strength of our business. To protect our brands’ status, we may need to make substantial expenditures to mitigate the impact of such threats.
We believe that maintaining and enhancing our brands in the countries in which we currently sell our products, and in new countries where we have limited brand recognition, is important to expanding our customer base. If we
31
are unable to maintain or enhance the strength of our brands in the countries in which we currently sell our products and in new countries, then our growth strategy could be adversely affected.
If we fail to compete effectively against our competitors, many of whom have greater resources than we have, our revenues and results of operations may be negatively affected.
Alternatives exist for Motiva Implants and for our other products, and we will likely face competition with respect to any planned products that we may seek to develop or commercialize in the future from major pharmaceutical companies, specialty pharmaceutical companies, medical device companies and biotechnology companies worldwide. There are several large pharmaceutical and biotechnology companies that currently market silicone breast implants. We also face competition from manufacturers of saline-filled breast implants, and we see emerging competition from non-implant breast augmentation techniques such as hyaluronic acid injection and novel fat grafting methodologies. Any of these may present competitive barriers to Motiva Implants.
Our leading competitors are large, multi-national companies with significant resources and capabilities. Three of these companies, Sientra, Inc., Mentor Worldwide LLC (a division of Johnson & Johnson), and Allergan plc (recently acquired by AbbVie Inc.), have conducted large prospective clinical studies that started in the United States in 2002, 2000 and 1998, respectively, and they use this data extensively to promote their products. This can put us at a disadvantage when promoting our products to physicians and patients, even outside the United States. In addition, the significant financial and staff resources and brand recognition that our competitors possess mean they may be able to compete with us regardless of the differentiating features of our products. If we are not successful in capturing market share, even outside the United States, or if physicians or patients do not perceive our products to be safer or more favorable, our revenues and/or our operating margins may be significantly impaired.
In addition, manufacturers of competitive products may reduce prices for their competing products in an effort to gain or retain market share and undermine the value proposition that Motiva Implants might otherwise be able to offer to customers. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization. These competitors may develop new technologies that are superior to our products or replace silicone.
Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties may compete with us in recruiting and retaining qualified technical and management personnel, establishing clinical study sites and patient registrations for clinical studies, as well as in acquiring technologies complementary to, or necessary for, our programs.
Pricing pressure from customers and our competitors may impact our ability to sell our products at prices necessary to support our current business strategies and future expansion.
The industry environment for silicone implants and complementary products in certain international markets is price sensitive. In these markets, or in the United States if we are successful in obtaining the required regulatory approval to sell in the U.S. market, our competitors may adopt aggressive pricing strategies to intensify the competitive pricing pressure for breast implants. If we are not successful in educating customers or third-party payors on the differentiation of our Motiva Implants as compared to our competitors’ products, customers may choose our competitors’ products. Additionally, as more competitors introduce products that compete with ours, we may face additional pricing pressure that would adversely impact our future results.
We expect to significantly increase the size of our organization; as a result, we may encounter difficulties in managing our growth, which could disrupt our operations and/or increase our net losses.
As of December 31, 2021, we had 746 employees. Unless it is necessary for us to continue to make reductions to our workforce as a cost management strategy due to the impact of the global COVID-19 pandemic on our business, over the next several years, we expect to experience significant growth in the number of our employees and the scope of our operations, principally in the areas of manufacturing, regulatory affairs, clinical and sales and marketing, and particularly as we prepare our operations in the anticipation of obtaining approval from the FDA to commercialize our Motiva Implants in the United States. We also intend to continue to improve our operational, financial and management controls, reporting systems and procedures, which may require additional personnel. Such growth could place a strain on our administrative and operational infrastructure, and/or our managerial
32
abilities, and we may not be able to make improvements to our management information and control systems in an efficient or timely manner. We may discover deficiencies in existing systems and controls.
Many of these employees will be in countries outside of our corporate headquarters, which adds additional complexity. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. We may not be able to effectively manage these activities. The physical expansion of our operations may lead to significant costs and may divert our management and business development resources. Future growth would impose significant added responsibilities on members of management, including:
•managing our clinical trials effectively, which we anticipate being conducted at numerous clinical sites;
•identifying, recruiting, maintaining, motivating and integrating additional employees with the expertise and experience we will require, in multiple countries;
•managing our internal development efforts effectively while complying with our contractual obligations to licensors, licensees, contractors and other third parties;
•managing additional relationships with various distributors, suppliers, and other third parties;
•improving our managerial, development, operational and finance reporting systems and procedures; and
•expanding our facilities.
Our failure to accomplish any of these tasks could prevent us from growing successfully. Any inability to manage growth could delay the execution of our business plans or disrupt our operations. We may also be exposed or subject to additional unforeseen or undisclosed liabilities as well as increased levels of indebtedness.
In certain large markets, we engage in direct sales efforts. We may fail to maintain and develop our direct sales force, and our revenues and financial outcomes could suffer as a result. Furthermore, our direct sales personnel may not effectively sell our products.
We have established a direct sales force for our business in Brazil, and we have implemented a direct sales strategy in several European countries. We have hired and will need to retain and motivate a significant number of sales and marketing personnel in order to support our anticipated growth in these countries. There is significant competition for quality personnel experienced in such activities, including from companies with greater financial resources than ours. If we are not successful in our efforts to continue recruiting, retaining, and motivating such personnel, we may not be able to increase our revenues, or we may increase our expenses in greater measure than our revenues, negatively impacting our operating results.
We are also working on creating a direct sales structure and strategy in certain markets. We are working to put in place the correct legal and business structures to comply with taxation and operational requirements. These structures may not ultimately be implemented or, if implemented, be successful or effective and may not be able to increase our revenues or improve our gross margins. In addition, our expenses or tax related costs may increase in greater measure than our revenues, negatively impacting our operating results.
We may be subject to substantial warranty or product liability claims or other litigation in the ordinary course of business that may adversely affect our business, financial condition and operating results.
We face an inherent risk of product liability exposure related to the sale of Motiva Implants and any planned products in clinical studies. The marketing, sale and use of Motiva Implants and our planned products could lead to the filing of product liability claims against us if someone alleges that our products failed to perform as designed or caused significant adverse events in patients. We may also be subject to liability for a misunderstanding of, or inappropriate reliance upon, the information we provide. If we cannot successfully defend ourselves against claims that Motiva Implants or our planned products caused injuries, we may incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
•decreased demand for any planned products we may develop;
•injury to our reputation and significant negative media attention;
•withdrawal of patients from clinical studies or cancellation of studies;
•significant costs to defend the related litigation and distraction to our management team;
33
•substantial monetary awards to plaintiffs;
•loss of revenue; and
•the inability to commercialize any products that we may develop.
We currently hold $25 million in product liability insurance coverage, which may not be adequate to cover all liabilities we may incur. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise.
Counterfeit products may be represented as ours, which could compete with our genuine products and may also expose us to risks associated with adverse events and product liability.
We routinely see counterfeit versions of our major competitor’s branded products in the marketplace, and we have recently become aware of potential counterfeiting of our Motiva Implants. This is particularly common in emerging markets, where sensitivity to price is higher and regulatory enforcement is under-resourced. These counterfeit products are typically manufactured with significantly lower quality than the products they are claimed to be, and in some cases may be manufactured with silicones that are not medical grade. They may expose patients to significant adverse event risks, and there is a risk that certain adverse events with counterfeit products may be attributed to our genuine products. This could reduce demand for our products, result in negative publicity, or otherwise impact our business and the price of our shares.
Negative publicity concerning our products or our competitors’ products, including due to product defects and any resulting litigation, could harm our reputation and reduce demand for silicone breast implants, either of which could adversely impact our financial results and/or share price.
The silicone breast implant industry has been the focus of significant regulatory and media scrutiny. Silicone breast implants were removed from the U.S. marketplace for a period in the 1990s and 2000s related to safety concerns. Certain patient advocacy groups exist to publicize real and perceived health risks associated with silicone breast implants and plastic surgery generally. Recently, some breast implant patients have begun to self-identify and report various symptoms that they believe are related to their breast implants; they refer to these symptoms as Breast Implant Illness, or BII, but BII is not an official medical diagnosis. Additionally, the activities of legislative bodies, regulatory agencies, physician organizations, and other groups may lead to publicity around the real and perceived risks to patients from silicone implants. The responses of potential patients, physicians, the news media, legislative and regulatory bodies and others to information about complications or alleged complications of our products or our competitors’ products, or products liability litigation against us or our competitors, could materially reduce market acceptance and patient demand for our products, or could, even in the absence of a change in demand, negatively impact our business and reputation and negatively impact our financial condition, results of operations or the market price of our common shares. In addition, activity of this type could result in an increase in the number or size of product liability claims, which would adversely affect our business, financial results, and/or the price of our shares.
Recent news coverage has called into question the long-term safety of breast implants and reports of breast implant-associated anaplastic large cell lymphoma (BIA-ALCL) linked to our competitors’ products which have led to regulatory actions regarding macrotextured devices in several countries and the worldwide recall of one of our competitor’s macrotextured implants and tissue expanders. These events may lead to a reduction in the demand for silicone breast implants and could adversely affect our business.
Women with breast implants have reported higher rates, as compared to the general population, of BIA-ALCL, an uncommon type of cancer affecting cells of the immune system. In January 2011, the FDA indicated that there was a possible association between certain saline and silicone gel-filled breast implants and higher rates of BIA-ALCL, with the causal links neither yet understood nor confirmed. In March 2015, France’s National Cancer Institute, or NCI, noted that there is a clearly established link between ALCL and certain breast implants, which is referred to as breast implant-associated ALCL, or BIA-ALCL. The NCI noted in that report that most of the reported cases occurred in women with textured implants.
In August 2017, the FDA updated its advisory on BIA-ALCL and subsequently requested all breast implant manufacturers to revise their physician and patient labeling with the most current information. The August 2017 update described BIA-ALCL as “rare” and stated “we have strengthened our understanding of this condition and
34
concur with the World Health Organization designation of BIA-ALCL as a rare T-cell lymphoma that can develop following breast implants. The exact number of cases remains difficult to determine due to significant limitations in world-wide reporting and lack of global implant sales data. At this time, most data suggest that BIA-ALCL occurs more frequently following implantation of breast implants with textured surfaces rather than those with smooth surfaces.” The FDA noted it does not recommend prophylactic breast implant removal in a patient without symptoms or other abnormalities.
In March 2018, the FDA further updated its advisory on BIA-ALCL stating “we are reporting that we are aware of 414 total cases of BIA-ALCL. Additionally, studies reported in medical literature estimate that the lifetime risk of developing BIA-ALCL for patients with textured breast implants ranges from 1 in 3,817 to 1 in 30,000.” The FDA noted that the update did not change the agency’s recommendation and that choosing to obtain a breast implant is a personal decision that patients and providers should make with the most complete information available. In the fourth quarter of 2018, following the non-renewal of its textured breast implant CE Mark licenses in Europe, Allergan plc suspended sales of textured breast implants in Europe and withdrew its remaining textured breast implants on the market within Europe.
On February 6, 2019, the FDA further reported that as of September 2018, the agency had received a total of 660 total medical device reports regarding BIA-ALCL cases since 2010. Of the 660 reports, the FDA’s analysis suggested that there are 457 unique cases of BIA-ALCL, including nine patient deaths. Additionally, on February 12, 2019, Health Canada confirmed that as of January 1, 2019, it had received reports of 22 confirmed and 22 suspected Canadian cases of BIA-ALCL and that it would be updating its safety review of BIA-ALCL in Spring 2019. In April 2019, the Agence Nationale de Securite du Medicament et des Produits de Sante, or ANSM, the regulatory authority in France, announced that 59 cases of BIA-ALCL had been reported in France since 2011 and banned several types of macrotextured and polyurethane implants linked to BIA-ALCL. Between February and September 2019, authorities from Australia, Colombia, Canada, South Korea and Singapore announced similar bans.
In July 2019, the FDA requested that Allergan plc recall its Biocell® textured implants in the U.S. market and Allergan subsequently announced the global recall of its Biocell® textured breast implants and tissue expanders. In the FDA announcement, it noted that it had reviewed 573 unique cases globally of BIA-ALCL, including 33 patient deaths, of which 12 of the 13 known deaths were attributed to Biocell® implants. The FDA further noted that it will continue to monitor the incidence of BIA-ALCL across other textured and smooth breast implants and tissue expanders as well as other devices intended for use in the breast. The FDA subsequently identified the recall as a Class I recall in September 2019 and stated that use of the recalled devices may cause serious injuries and death. As the BIA-ALCL risk continues to become more highly publicized, this could have a significant negative impact on demand for breast implants globally, including our Motiva Implants.
In September 2020, the FDA released finalized guidance on breast implant labeling recommendations, including the addition of a boxed warning, a patient decision checklist, material and device descriptions, implant rupture screening recommendations and a patient device card. In October 2021, the FDA took several additional actions to strengthen breast implant risk communication, including restricting the sale and distribution of breast implants to only health care providers and facilities that provide information to patients using the patient decision checklist. The FDA also approved new labeling for all legally marketed breast implants that includes a boxed warning, a patient decision checklist, updated silicone gel-filled breast implant rupture screening recommendations, a device description with a list of specific materials, and a patient device card.
In August 2020, the FDA updated its analysis of medical device reports of breast implant illness and breast implant associated lymphoma. In this update, the FDA updated the table on the agency’s BIA-ALCL webpage to include a total of 733 unique cases and 36 patient deaths globally as of January 5, 2020, which reflect an increase of 160 new cases and 3 deaths since the early-July 2019 update.
We do not produce the types of rough textured implants that have been involved in these reports. To date, no cases of BIA-ALCL have been reported in patients with Motiva Implants. Furthermore, there have been no reported cases of BIA-ALCL in patients with smooth implants with no history of previously having a textured device. Future clinical studies or clinical experience may indicate that breast implants expose potentially genetically predisposed patients to greater risks of BIA-ALCL, which may reduce demand for silicone implants generally, expose us to product liability claims, as well as to class actions and other lawsuits. These impacts may occur in the absence of any specific linkage with our products. Moreover, if cases of BIA-ALCL or other complications are discovered in the future and/or are reported in patients with Motiva Implants, we could be subject to mandatory product recalls, suspension or withdrawal of our regulatory licensure for sale in one or more
35
countries, and significant legal liability. Any of these may have an adverse effect on our business or operating results, or a negative impact on our share price.
The loss of members of our executive management team or other employees, or other turnover in our management team, could adversely affect our business.
Our success in implementing our business strategy depends largely on the skills, experience and performance of members of our executive management team and other key employees, including Juan José Chacón Quirós, our Chief Executive Officer, Roberto de Mezerville, our Chief Technology Officer, Rajbir Denhoy, our Chief Financial Officer, and Pratip Dastidar, our Head of Global Operations. The collective efforts of each of these persons, and others working with them as a team, are critical as we continue to develop our tests and technologies and pursue our research and development and sales programs. In addition, we have experienced significant changes in our executive leadership in the past twelve months, including in our Chief Financial Officer and Chief Operating Officer positions. As a result of the difficulty in locating qualified new management and other key employees, the loss or incapacity of existing members of our executive management team could adversely affect our operations. If we were to lose one or more key employees, we could experience difficulties in finding qualified successors, competing effectively, developing our technologies and implementing our business strategy. In addition, changes to strategic or operating goals, which can often times occur with the appointment of new executives and directors, can create uncertainty, may negatively impact our ability to execute quickly and effectively, and may ultimately be unsuccessful. Executive leadership transition periods are often difficult as the new executives gain detailed knowledge of our operations, and friction can result from changes in strategy and management style. Management turnover inherently causes some loss of institutional knowledge, which can negatively affect strategy and execution. We do not have “key person” life insurance on our senior executives, and the loss of any of the key team members would have a negative impact to our business and financial results. In addition, the job market in Costa Rica and other locations in which we operate has recently become more competitive and we are competing for talent with major multinational corporations which have significantly more resources than us, and we may find new difficulties in retaining our most talented employees.
In addition, we rely on collaborators, consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our collaborators, consultants and advisors are generally employed by employers other than us and may have commitments under agreements with other entities that may limit their availability to us.
We continue to incur significant costs as a result of operating as a public company, and our management is required to devote substantial time to new compliance initiatives.
As a public company, we incur significant legal, accounting and other expenses that we did not incur as a private company. We are subject to the reporting requirements of the Exchange Act, the other rules and regulations of the Securities and Exchange Commission, or SEC, and the rules and regulations of Nasdaq. The expenses that are required in order to adequately prepare for being a public company are material, and compliance with the various reporting and other requirements applicable to public companies require considerable time and attention of management. For example, the Sarbanes-Oxley Act and the rules of the SEC and national securities exchanges have imposed various requirements on public companies, including requiring establishment and maintenance of effective disclosure and financial controls. Our management and other personnel devote a substantial amount of time to these compliance initiatives. These rules and regulations continue to increase our legal and financial compliance costs and make some activities more time-consuming and costly. For example, these rules and regulations make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits on coverage or incur substantial costs to maintain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified personnel to serve on our Board of Directors, our board committees, or as executive officers.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal control over financial reporting and disclosure controls and procedures. In particular, we must perform system and process evaluation and testing of our internal control over financial reporting to allow management to report on the effectiveness of our internal control over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act. In addition, we are required to have our independent registered public accounting firm attest to the effectiveness of our internal control over financial reporting. Our compliance with Section 404 of the Sarbanes-Oxley Act requires that we incur substantial accounting expense and expend significant management efforts. We currently do not have an internal audit group, and we will need to hire additional accounting and financial staff with appropriate public company
36
experience and technical accounting knowledge. If we are not able to comply with the requirements of Section 404 in a timely manner, or if we or our independent registered public accounting firm identify deficiencies in our internal control over financial reporting that are deemed to be material weaknesses, the market price of our shares could decline and we could be subject to sanctions or investigations by Nasdaq, the SEC or other regulatory authorities, which would require additional financial and management resources.
Our ability to successfully implement our business plan and comply with Section 404 requires us to be able to prepare timely and accurate financial statements. We expect that we will need to continue to improve existing, and implement new operational and financial systems, procedures and controls to manage our business effectively. Any delay in the implementation of, or disruption in the transition to, new or enhanced systems, procedures or controls, may cause our operations to suffer and we may be unable to conclude that our internal control over financial reporting is effective and to obtain an unqualified report on internal controls from our auditors as required under Section 404 of the Sarbanes-Oxley Act. This, in turn, could have an adverse impact on trading prices for our common shares, and could adversely affect our ability to access the capital markets.
We have made multiple acquisitions in the past, and in the future we may acquire other businesses or form joint ventures or make investments in other companies or technologies. If we are not successful in integrating these businesses, as well as identifying and controlling risks associated with the past operations of these businesses, we may incur significant costs, receive penalties or other sanctions from various regulatory agencies, and/or incur significant diversions of management time and attention.
We believe our business growth will be enhanced if we continually seek opportunities to enhance and broaden our product offerings. As part of our business strategy, we may pursue acquisitions or licenses of assets, or acquisitions of businesses. We also may pursue strategic alliances and joint ventures that leverage our core technology and industry experience to expand our product offerings or sales and distribution resources. We have acquired companies and/or assets and licensed assets in a variety of countries, including Brazil and several European countries.
We may do more of these types of transactions in the future and may also form strategic alliances and joint ventures. We may not be able to find suitable partners or acquisition candidates, and we may not be able to complete such transactions on favorable terms, if at all. If we make any acquisitions, we may not be able to integrate these acquisitions successfully into our existing business, and we could assume unknown or contingent liabilities. Any future acquisitions also could result in significant write-offs or the incurrence of debt and contingent liabilities, any of which could have an adverse effect on our financial condition, results of operations and cash flows. Integration of an acquired company may also disrupt ongoing operations and require management resources that would otherwise focus on developing our existing business. We may experience losses related to investments in other companies, which could have a negative effect on our results of operations. We may not identify or complete these transactions in a timely manner, on a cost-effective basis, or at all, and we may not realize the anticipated benefits of any acquisition, license, strategic alliance or joint venture. To finance such a transaction, we may choose to issue common shares as consideration, which would dilute the ownership of our shareholders. If the price of our common shares is low or volatile, we may not be able to acquire other companies or fund a joint venture project using our shares as consideration. Alternatively, it may be necessary for us to raise additional funds for acquisitions through public or private financings. Additional funds may not be available on terms that are favorable to us, or at all.
We do not know whether we will be able to successfully integrate any acquired business, product or technology. The success of any given acquisition may depend on our ability to retain any key employees related thereto, and we may not be successful at retaining or integrating such key personnel. Integrating any business, product or technology we acquire could be expensive and time-consuming, disrupt our ongoing business, impact our liquidity, and/or distract our management. If we are unable to integrate any acquired businesses, products or technologies effectively, our business may suffer. Whether as a result of unsuccessful integration, unanticipated costs, including those associated with assumed liabilities and indemnification obligations, negative accounting impact, or other factors, we may not realize the economic benefits we anticipate from acquisitions. In addition, any amortization or charges resulting from the costs of acquisitions could increase our expenses.
Unfavorable global economic conditions could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy and in the global financial markets. The global financial crisis caused extreme volatility and disruptions in the capital and
37
credit markets. A severe or prolonged economic downturn, could result in a variety of risks to our business, including general economic pressure on our customers’ patients. Elective aesthetic procedures, including breast augmentation, are typically not covered by insurance and are less of a priority than other items for those patients that have lost their jobs, are furloughed, have reduced work hours or have to allocate their cash to other priorities. A weak or declining economy could also strain our manufacturers or suppliers, possibly resulting in supply disruption, or cause our customers or distributors to delay making payments for our products. Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the economic climate and financial market conditions could adversely affect our business.
We have significant exposure to the economic and political situations in emerging market countries, and developments in these countries could materially impact our financial results, or our business more generally.
Many of the countries in which our products are sold are emerging markets. Our global growth strategy contemplates the expansion of our existing sales activities in Latin America, Europe, the Middle East, and Asia-Pacific region as well as North America. Our exposure to emerging markets has increased in recent years, as have the number and importance of our distributor arrangements. Economic and political developments in Brazil and other emerging markets, including economic crises, currency inflation, or political instability, have had in the past, and may have in the future, a material adverse effect on our financial condition and results of operations. Moreover, as these markets continue to grow, competitors may seek to enter these markets and existing market participants will likely try to aggressively protect or increase their market shares. Increased competition may result in price reductions, reduced margins and our inability to gain or hold market share, which could have an adverse effect on our financial condition and results of operations.
Our results of operations could be affected by fluctuations in currency rates.
We present our results of operations in U.S. dollars, which is our reporting currency. However, as of December 31, 2021, the majority of our revenues are denominated in currencies other than the U.S. dollar - primarily the euro, and the Brazilian real, and the British pound. As of December 31, 2021, the majority of our expenses are denominated in U.S. dollars or in Costa Rican colones, which are linked to the U.S. dollar. In the future, we expect to have significant revenues and expenses denominated in these non-U.S. currencies. As such, unfavorable fluctuations in currency exchange rates could have an adverse effect on our results of operations.
Because our consolidated financial statements are presented in U.S. dollars, we must translate revenues, expenses and income, as well as assets and liabilities, into U.S. dollars at exchange rates in effect during or at the end of each reporting period. Therefore, changes in the value of the U.S. dollar in relation to the British pound, the euro, and the Brazilian real will affect our revenues, operating income and the value of balance sheet items originally denominated in other currencies. These changes would cause our growth in consolidated earnings stated in U.S. dollars to be higher or lower than our growth in local currency when compared against other periods. We do not currently engage in currency hedging arrangements to protect us from fluctuations in the exchange rates of the euro and other currencies in relation to the U.S. dollar (and/or from inflation of such currencies), and we may be exposed to material adverse effects from such movements. We cannot predict any future trends in rates of inflation or exchange rates of other currencies against the U.S. dollar, and there can be no assurance that any contractual provisions will offset their impact, or that any future currency hedging activities will be successful.
Continued international expansion of our business will expose us to business, regulatory, political, operational, financial and economic risks associated with doing business internationally.
Our products are commercially available in more than 80 countries, and we operate subsidiaries in the United States, Costa Rica, Brazil, and several European countries. Our business strategy contemplates continued international expansion, including partnering with medical device distributors, and introducing Motiva Implants and other planned products outside the United States. The sale and shipment of our products internationally, as well as the purchase of components from international sources, subjects us to potential trade, import and export, and customs regulations and laws.
Compliance with these regulations and laws is costly and exposes us to penalties for non-compliance. Any failure to comply with applicable legal and regulatory obligations could impact us in a variety of ways that include, but are not limited to, significant criminal, civil and administrative penalties, including imprisonment of individuals, fines and penalties, denial of export or import privileges, seizure of shipments, restrictions on certain business activities
38
and exclusion or debarment from government contracting. Also, the failure to comply with applicable legal and regulatory obligations could result in the disruption of our shipping, marketing and sales activities.
In addition, several of the countries in which we sell our products or conduct our operations are, to some degree, subject to political, economic or social instability. Doing business in Costa Rica and other countries outside the United States involves a number of other risks, including:
•compliance with the free zone regime regulations under which the manufacturing sites operate;
•different regulatory requirements for device approvals in international markets;
•multiple, conflicting and changing laws and regulations such as tariffs and tax laws, export and import restrictions, employment laws, regulatory requirements and other governmental approvals, permits and licenses;
•potential failure by us or our distributors to obtain and/or maintain regulatory approvals for the sale or use of our products in various countries;
•difficulties in managing global operations;
•logistics and regulations associated with shipping products, including infrastructure conditions and transportation delays;
•limits on our ability to penetrate international markets if our distributors do not execute successfully;
•governmental price controls, differing reimbursement regimes and other market regulations;
•financial risks, such as longer payment cycles, difficulty enforcing contracts and collecting accounts receivable, and exposure to currency exchange rate fluctuations;
•reduced protection for intellectual property rights, or lack of them in certain jurisdictions, forcing more reliance on our trade secrets, if available;
•economic weakness, political and economic instability, including wars, terrorism and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions;
•the British exit from the EU, including with respect to its effect on the value of the British pound relative to other currencies;
•failure to comply with the Foreign Corrupt Practices Act, including its books and records provisions and its anti-bribery provisions, by maintaining accurate information and control over sales activities and distributors’ activities;
•failure to comply with restrictions on the ability of companies to do business in foreign countries, including restrictions on foreign ownership of telecommunications providers imposed by the U.S. Office of Foreign Assets Control;
•unexpected changes in tariffs, trade barriers and regulatory requirements;
•compliance with tax, employment, immigration and labor laws;
•taxes, including withholding of payroll taxes;
•currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country;
•workforce uncertainty in countries where labor unrest is more common than in the United States;
•production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and
•business and shipping interruptions resulting from natural or other disasters including earthquakes, volcanic activity, hurricanes, floods and fires.
Any of these risks, if encountered, could harm our future international expansion and operations and, consequently, have an adverse effect on our financial condition, results of operations and cash flows
39
Any disruption at our existing facilities could adversely affect our business and operating results.
Our headquarters are located in Costa Rica, and all of our main manufacturing activities are conducted in two ISO-13485 and GMP compliant manufacturing facilities in Costa Rica through Establishment Labs, S.A. Despite our efforts to maintain and safeguard our manufacturing facilities, including acquiring insurance and adopting maintenance and health and safety protocols, vandalism, terrorism or a natural or other disaster, such as earthquake, volcanic activity, fire or flood, could damage or destroy our inventory of finished goods, cause substantial delays in our operations and manufacturing, result in the loss of key information and cause us to incur additional expenses. Our insurance may not cover our losses in any particular case. In addition, regardless of the level of insurance coverage, damage to our facilities may have an adverse effect on our business, financial condition and results of operations.
Fluctuations in insurance costs and availability, and future insurance requirements could adversely affect our profitability or our risk management profile.
We hold a number of insurance policies, including product liability insurance, directors’ and officers’ liability insurance, general liability insurance, property insurance and workers’ compensation insurance. If the costs of maintaining adequate insurance coverage increase significantly in the future, our operating results could be adversely affected. Likewise, if any of our current insurance coverage should become unavailable to us or become economically impractical, we would be required to operate our business without indemnity from commercial insurance providers. If we operate our business without insurance, we could be responsible for paying claims or judgments against us that would have otherwise been covered by insurance, which would adversely affect our results of operations or financial condition.
Risks Related to Manufacturing and Other Third-Party Relationships
Our operations involve hazardous materials and we and third parties with whom we contract must comply with environmental laws and regulations, which can be expensive and restrict how we do business, and could expose us to liability if our use of such hazardous materials causes injury.
Our manufacturing processes currently require the controlled use of potentially harmful chemicals, including highly flammable solvents. We cannot eliminate the risk of accidental contamination or injury to employees or third parties from the use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for any resulting damages, and any liability could exceed our resources or any applicable insurance coverage we may have. Additionally, we are subject to, on an ongoing basis, federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. These are particularly stringent in California, where NuSil, one of our key suppliers, is located. The cost of compliance with these laws and regulations may become significant and could have an adverse effect on our financial condition, results of operations and cash flows. In the event of an accident or if we otherwise fail to comply with applicable regulations, we could lose our permits or approvals or be held liable for damages or penalized with fines.
We rely on third parties to conduct certain components of our clinical studies, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such studies, which could interfere with or delay our ability to get regulatory approval or commercialize our products.
We rely on third parties, such as contract research organizations, or CROs, clinical data management organizations, medical institutions and clinical investigators, to perform various functions for our clinical trials. Our reliance on these third parties for clinical development activities reduces our control over these activities but does not relieve us of our responsibilities. We remain responsible for ensuring that each of our clinical studies is conducted in accordance with the general investigational plan and protocols for the study. Moreover, the International Council for Harmonization, or ICH, and the FDA require us to comply with standards, commonly referred to as good clinical practices, for conducting, recording and reporting the results of clinical studies to ensure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of patients in clinical studies are protected. Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical studies in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, regulatory approvals for our planned products and will not be able to, or may be delayed in our efforts to, successfully commercialize our planned products.
40
We rely on a single-source, third-party supplier for medical-grade long-term implantable silicone, which is the primary raw material used in our Motiva Implants. If this supplier were to increase prices for this raw material over time or experience interruptions in its ability to supply us with this raw material, our business, financial condition and results of operations could be adversely affected.
We rely on NuSil, as the sole supplier of medical-grade silicone used in our Motiva Implants as well as other products that we manufacture under contract to other customers. To our knowledge, NuSil is the only supplier of such raw materials with the appropriate filings with the FDA and other regulatory bodies to enable the manufacturing of products with our requirements. NuSil supplies our major competitors with raw material as well, and at least two of these are larger-volume customers of NuSil than we are.
If NuSil becomes unable or unwilling to supply sufficient quantities of medical-grade silicone of the specifications required for our products, we may not be able to replace this supply source quickly, or at all. Similarly, they may become unable or unwilling to manufacture our needed raw materials in compliance with regulatory requirements, or their manufacturing facilities may not be able to maintain compliance with regulatory requirements. Any replacement supplier would have to be qualified with the relevant regulatory authorities, which is an expensive and time-consuming process during which we may experience an interruption in our manufacturing operations. We may also be unsuccessful in negotiating favorable terms with such a supplier. Any of these contingencies would likely affect the financial results of our operations and may have a negative impact on our share price. In particular, if we are not able to establish a replacement vendor for our medical-grade silicone, we would be unable to manufacture our Motiva Implants as well as other products that we manufacture under contract to other customers until such time as a replacement vendor is identified, which would likely significantly affect the financial results of our operations and have a significantly negative impact on our share price.
We are in the process of renegotiating our current supply agreement with NuSil, which is due to expire on March 31, 2022. There can be no assurance that NuSil will agree to continue to supply us with medical-grade silicone following the expiration of our contract on terms that are acceptable to us, or at all. This would have a material adverse effect on our business, financial condition, and results of operations for the reasons set forth above.
In addition, our relationship with NuSil involves other risks, including but not limited to the following:
•it may not be able, or willing, to manufacture silicone raw materials with our agreed-upon specifications;
•it may not be able, or willing, to manufacture our needed raw materials in compliance with regulatory requirements, or our its manufacturing facilities may not be able to maintain compliance with regulatory requirements;
•it may not be able to supply sufficient quantities of each raw material quickly enough for us to respond to rapid increases in demand;
•it may unintentionally convey information to our competitors that is helpful in understanding our proprietary compositions and other trade secrets of our manufacturing processes;
•we may be subject to price fluctuations if we fail to meet certain minimum order requirements, or if our existing contract expires or is renegotiated;
•it may lose access to critical services and components, resulting in interruption in manufacture or shipment of medical-grade silicone;
•its facilities may be affected by earthquakes, wildfires, mud slides or other natural disasters, which could delay or impede production of our raw materials;
•we may be required to obtain regulatory approvals related to any change in our supply chain;
•NuSil may wish to discontinue supply of products to us due to its existing relationships with our competitors;
•NuSil may stop supply and claim ownership of intellectual property on materials associated with future products; and
•NuSil or its parent entity may encounter financial or other hardships unrelated to our demand for products, which could negatively impact their ability to fulfill our orders and support our regulatory approvals.
41
Various factors outside our direct control, including the reliance on single-source suppliers, may adversely affect manufacturing and supply of our Motiva Implants and other products.
We currently manufacture Motiva Implants at our facilities in the Coyol Free Zone, Alajuela, Costa Rica, under the multi-country MDSAP protocol. Our Qid Safety Technology microtransponders are manufactured by contract manufacturers with final testing and packaging at a manufacturing supplier facility in Regensburg, Germany, with additional inspection of the units at our facilities in Coyol, Costa Rica, prior to approval for inclusion in Motiva Implants. If demand for our current products and our planned products increases more rapidly than we anticipate, or if we secure regulatory approval to commercialize our products in additional geographies, we will need to either expand our manufacturing capabilities or outsource to other manufacturers. The manufacture of these products in compliance with ISO standards and the FDA’s regulations requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of medical device products often encounter difficulties in production, including difficulties with production costs and yields, quality control, quality assurance testing, shortages of qualified personnel, as well as compliance with strictly enforced FDA requirements, other federal and state regulatory requirements, and foreign regulations.
We currently purchase components for the Qid Safety Technology microtransponders under purchase orders and do not have long-term contracts with most of the suppliers of the materials included in these products. We rely on NuSil Technology, LLC, or NuSil, as the sole supplier of medical-grade silicone used in our Motiva Implants as well as other products that we manufacture under contract to other customers. See the risk factor below titled “We rely on a single-source, third-party supplier for medical-grade silicone, which is the primary raw material used in these products. If this supplier were to increase prices for these raw materials over time or experience interruptions in their ability to supply us with this raw material, our business, financial condition and results of operations could be adversely affected.” In addition, the suppliers of certain packaging components and the surgical tools that we sell with Motiva Implants, including the cannulas, retractors, and insertion sleeves, are all purchased by us from single-source suppliers.
If our single-source and other suppliers were to delay or stop producing our components, or if the prices they charge us were to increase significantly, or if they elected not to sell to us at all or on commercially reasonable terms, we would need to identify and initiate relationships with alternative suppliers, if possible. We could experience delays in manufacturing our products or the interruption of the availability of Motiva Implants or our other products for sale, while finding another acceptable supplier, which would impact our business, financial condition and results of operations. Even if such alternative suppliers are available on commercially reasonable terms, the changes could also result in increased costs associated with qualifying the new materials and in increased operating costs. Further, any prolonged disruption in a supplier’s operations could have a significant negative impact on our ability to manufacture and deliver products in a timely manner and as a result, our business, financial condition and results of operations could be adversely affected.
The manufacturing, sterilization and distribution of our Motiva Implants and other products are technically challenging. Changes that our suppliers may make, or additional requirements from regulatory agencies, are outside of our direct control and can have an impact on our processes, on quality, and on the successful delivery of products to our customers. Mistakes and mishandling are not uncommon and can affect supply and delivery. Some of these risks include:
•failure to complete sterilization on time or in compliance with the required regulatory standards;
•transportation and import and export risk, particularly given the global nature of our supply and distribution chains;
•delays in analytical results or failure of analytical techniques that we depend on for quality control and release of products;
•natural or other disasters, labor disputes, financial distress, lack of raw material supply, issues with facilities and equipment or other forms of disruption to business operations affecting our manufacturer or its suppliers;
•latent defects that may become apparent after products have been released and that may result in a recall of such products;
•contamination of our raw materials or manufactured products; and
•inclusion of vendors of raw materials not in compliance with ISO-13485 requirements.
42
As referenced above in this risk factor, some of the components used in our Motiva Implants and our other products are currently single-sourced, and substitutes for these components might not be obtained easily or may require substantial redesign or manufacturing modifications related to our specifications or due to regulatory requirements. Any significant problem experienced by one of our single-source suppliers may result in a delay or interruption in the supply of components or products to us because the number of third-party manufacturers with the necessary manufacturing and regulatory expertise and facilities is limited and certification of a new supplier may be complex and time consuming. Any delay or interruption would likely lead to a delay or interruption in our manufacturing or distribution operations and/or adversely affect our ability to sell Motiva Implants. The inclusion of substitute components or products must meet our specifications and could require us to qualify the new supplier with the appropriate regulatory authorities. The added time and cost to arrange for alternative suppliers could have a material adverse effect on our business. New manufacturers of any current or planned product would be required to qualify under applicable regulatory requirements and would need to have sufficient rights under applicable intellectual property laws to the design and method of manufacturing the planned product. Obtaining the necessary FDA or international approvals or other qualifications under applicable regulatory requirements and ensuring non-infringement of third-party intellectual property rights could result in a significant interruption of supply and could require the new manufacturer to bear significant additional costs that may be passed on to us.
A substantial proportion of our sales are through exclusive distributors, and we do not have direct control over the efforts these distributors may use to sell our products. If our relationships with these third-party distributors deteriorate, or if these third-party distributors fail to sell our products or engage in activities that harm our reputation, or fail to adhere to medical device regulations, our financial results may be negatively affected.
Historically, our sales model has been to sell primarily through distributors rather than through our own sales force, with the notable exception of Brazil and several European countries where we are selling directly, but, in the future, we may utilize a hybrid sales model that includes both distributors and a direct sales effort. We believe that our reliance on distributors improves the economics of our business, as we do not carry the high fixed costs of a direct sales force in many of the countries in which our Motiva Implants are sold. If we are unable to maintain or enter into such distribution arrangements on acceptable terms, or at all, we may not be able to successfully commercialize our products in certain countries. Furthermore, distributors can choose the level of effort that they apply to selling our products relative to others in their portfolio. The selection, training, and compensation of a distributors’ sales personnel are within their control rather than our own and may vary significantly in quality from distributor to distributor.
In addition, although our contract terms require our distributors to comply with all applicable laws regarding the sale of our products, including anti-competition, anti-money laundering, and sanctions laws, we may not be able to ensure proper compliance. If our distributors fail to effectively market and sell our products in full compliance with applicable laws, our results of operations and business may suffer.
Risks Related to Intellectual Property and Data Security
If we are unable to protect the confidentiality of our trade secrets, the value of our technology could be materially adversely affected, harming our business and competitive position.
In addition to our patented technology and products, we rely upon confidential proprietary information, including trade secrets, unpatented know-how, technology and other proprietary information, to develop and maintain our competitive position. Any disclosure to or misappropriation by third parties of our confidential proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, thus eroding our competitive position in the market. We seek to protect our confidential proprietary information, in part, by confidentiality agreements with our employees and our collaborators and consultants. We also have agreements with our employees and selected consultants that obligate them to assign their inventions to us. These agreements are designed to protect our proprietary information, however, we cannot be certain that our trade secrets and other confidential information will not be disclosed or that competitors will not otherwise gain access to our trade secrets, or that technology relevant to our business will not be independently developed by a person that is not a party to such an agreement. Furthermore, if the employees, consultants or collaborators that are parties to these agreements breach or violate the terms of these agreements, we may not have adequate remedies for any such breach or violation, and we could lose our trade secrets through such breaches or violations. Further, our trade secrets could be disclosed, misappropriated or otherwise become known or be independently discovered by our competitors. In addition, intellectual property laws in foreign countries may not
43
protect trade secrets and confidential information to the same extent as the laws of the United States. If we are unable to prevent disclosure of the intellectual property related to our technologies to third parties, we may not be able to establish or maintain a competitive advantage in our market, which would harm our ability to protect our rights and have an adverse effect on our business.
If we are not able to obtain and maintain intellectual property protection for our products and technologies, or if the scope of our patents is not sufficiently broad, we may not be able to effectively maintain our market leading technology position.
Our success depends in large part on our ability to obtain and maintain patent and other intellectual property protection in the United States and in other countries with respect to our proprietary technology and products.
The patent position of medical device and diagnostic companies generally is highly uncertain and involves complex legal and factual questions for which legal principles remain unresolved. In recent years, patent rights have been the subject of significant litigation. As a result, the issuance, scope, validity, enforceability and commercial value of the patent rights we rely on are highly uncertain. Pending and future patent applications may not result in patents being issued which protect our technology or products or which effectively prevent others from commercializing competitive technologies and products. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of the patents we rely on or narrow the scope of our patent protection. The laws of other countries may not protect our rights to the same extent as the laws of the U.S. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot be certain that we were the first to make the inventions claimed in our patents or pending patent applications, or that we or were the first to file for patent protection of such inventions.
Even if the patent applications we rely on issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner. The issuance of a patent is not conclusive as to its scope, validity or enforceability, and the patents we rely on may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in patent claims being narrowed, invalidated or held unenforceable, which could limit our ability to stop or prevent us from stopping others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. Given the amount of time required for the development, testing and regulatory review of new planned products, patents protecting such products might expire before or shortly after such products are commercialized. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours or otherwise provide us with a competitive advantage.
We may not be able to protect or enforce our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on all of our planned products throughout the world may be prohibitively expensive to us. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection but where enforcement is not as strong as in the United States. These products may compete with our products in jurisdictions where we do not have any issued patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing. Many companies have encountered significant problems in protecting and defending intellectual property rights in international jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
We may become involved in legal proceedings to protect or enforce our intellectual property rights, which could be expensive, time consuming, or unsuccessful.
Competitors may infringe or otherwise violate the patents we rely on, or our other intellectual property rights. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. Any claims that we assert against perceived infringers could also provoke these parties to
44
assert counterclaims against us alleging that we infringe their intellectual property rights. In addition, in an infringement proceeding, a court may decide that a patent we are asserting is invalid or unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that the patents we are asserting do not cover the technology in question. An adverse result in any litigation proceeding could put one or more patents at risk of being invalidated or interpreted narrowly. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
Interference or derivation proceedings provoked by third parties or brought by the U.S. Patent and Trademark Office, or USPTO, or any other patent authority may be necessary to determine the priority of inventions or other matters of inventorship with respect to patents and patent applications. We may become involved in proceedings, including oppositions, interferences, derivation proceedings, inter partes reviews, patent nullification proceedings, or re-examinations, challenging our patent rights or the patent rights of others, and the outcome of any such proceedings are highly uncertain. An adverse determination in any such proceeding could reduce the scope of, or invalidate, important patent rights, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights. Our business also could be harmed if a prevailing party does not offer us a license on commercially reasonable terms, if any license is offered at all. Litigation or other proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may also become involved in disputes with others regarding the ownership of intellectual property rights. If we are unable to resolve these disputes, we could lose valuable intellectual property rights.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses and could distract our technical or management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the market price of our common shares. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. Uncertainties resulting from the
45
initiation and continuation of intellectual property litigation or other proceedings could have an adverse effect on our ability to compete in the marketplace.
The medical device industry is characterized by patent litigation and we could become subject to litigation that could be costly, result in the diversion of management's time and efforts, require us to pay damages or prevent us from marketing our existing or future products.
Patent litigation is prevalent in the medical device and diagnostic sectors. Our commercial success depends in part upon our ability and that of our distributors, contract manufacturers, and suppliers to manufacture, market and sell our planned products, and to use our proprietary technologies without infringing, misappropriating or otherwise violating the proprietary rights or intellectual property of third parties. We may become party to, or be threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our products and technology. Third parties may assert infringement claims against us based on existing or future intellectual property rights. If we are found to infringe a third-party’s intellectual property rights, we could be required to obtain a license from such third party to continue developing and marketing our products and technology. We may also elect to enter into such a license in order to settle pending or threatened litigation. However, we may not be able to obtain any required license on commercially reasonable terms, or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us and could require us to pay significant royalties and other fees. We could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, we could be found liable for monetary damages. A finding of infringement could prevent us from commercializing our planned products in commercially important territories, or force us to cease some of our business operations, which could harm our business. Many of our employees were previously employed at, and many of our current advisors and consultants are employed by, universities or other biotechnology, medical device or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees, advisors and consultants do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we, or these employees, have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such employee’s former employer. These and other claims that we have misappropriated the confidential information or trade secrets of third parties can have a similar negative impact on our business to the infringement claims discussed above.
Even if we are successful in defending against intellectual property claims, litigation or other legal proceedings relating to such claims may cause us to incur significant expenses, and could distract our technical and management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common shares. Such litigation or proceedings could substantially increase our operating losses and reduce our resources available for development activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their substantially greater financial resources. Uncertainties resulting from the initiation and continuation of litigation or other intellectual property related proceedings could have a material adverse effect on our ability to compete in the marketplace.
Obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents or applications will be due to be paid by us to the USPTO and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents or applications. The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. In many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to use our technologies and this circumstance would have a material adverse effect on our business.
46
If we fail to comply with our obligations in our intellectual property agreements, we could lose intellectual property rights that are important to our business.
We are a party, and expect to become party in the future, to certain intellectual property agreements that impose various diligence, milestone payment, royalty, insurance and other obligations on us. If we fail to comply with these obligations, any licensor may have the right to terminate such agreements, in which event we may not be able to develop and market any product that is covered by such agreements. Termination of such agreements, or reduction or elimination of our rights under such agreements, may result in our having to negotiate new or reinstated arrangements on less favorable terms, or our not having sufficient intellectual property rights to operate our business. The occurrence of such events could harm our business and financial condition.
The risks described elsewhere in this Annual Report on Form 10-K pertaining to our intellectual property rights also apply to any intellectual property rights that we may license, and any failure by us or any future licensor to obtain, maintain, defend and enforce these rights could have a material adverse effect on our business.
Our internal computer systems, or those used by third parties which we rely on, may fail or suffer security breaches.
Despite the implementation of security measures, our internal computer systems, or those used by third parties which we rely on, are vulnerable to damage from computer viruses and unauthorized access, malware, natural disasters, fire, terrorism, war and telecommunication, electrical failures, cyber-attacks or cyber-intrusions over the Internet, attachments to emails, persons inside our organization, or persons with access to systems inside our organization. The risk of a security breach or disruption, particularly through cyber-attacks or cyber intrusion, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. While we have not experienced any such material system failure or security breach to our knowledge to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations. For example, the loss of data from completed, ongoing or future studies could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development and commercialization of our current and future products could be delayed. In addition, any such disruption or security breach could harm our reputation, erode customer confidence in the effectiveness of our security measures, and negatively impact our ability to attract new customers.
Our failure to adequately protect personal information in compliance with evolving legal requirements could harm our business.
In the ordinary course of our business, we collect and store sensitive data, including legally protected patient health information, credit card information and personally identifiable information. We collect this kind of information on our customers for purposes of servicing potential warranty claims and for post-marketing safety vigilance. These data protection and privacy-related laws and regulations are evolving and may result in ever-increasing regulatory and public scrutiny and escalating levels of enforcement and sanctions.
There are a number of state, federal and international laws protecting the privacy and security of health information and personal data. As part of the American Recovery and Reinvestment Act 2009, or ARRA, Congress amended the privacy and security provisions of the Health Insurance Portability and Accountability Act, or HIPAA. HIPAA imposes limitations on the use and disclosure of an individual’s protected health information by certain health care providers, health care clearinghouses, and health insurance plans, collectively referred to as covered entities, that involve the creation, use, maintenance or disclosure of protected health information. The HIPAA amendments also impose compliance obligations and corresponding penalties for non-compliance on individuals and entities that provide services to health care providers and other covered entities, collectively referred to as business associates. Most recently, on December 10, 2020, HHS issued a Notice of Proposed Rulemaking, which if finalized, would make changes to some of HIPAA’s regulatory requirements, which would impact us, to the extent we are a business associate. ARRA also made significant increases in the penalties for improper use or disclosure of an individual’s protected health information under HIPAA and extended enforcement authority to state attorneys general. The amendments also create notification requirements for individuals whose protected health information has been inappropriately accessed or disclosed, notification requirements to federal regulators and in some cases, notification to local and national media. Notification is not required under HIPAA if the health information that is improperly used or disclosed is deemed secured in accordance with encryption or other standards developed by the U.S. Department of Health and Human Services, or HHS. Most states have laws requiring notification of affected individuals and state regulators in the event of a breach of personal information,
47
which is a broader class of information than the protected health information protected by HIPAA. Many state laws impose significant data security requirements, such as encryption or mandatory contractual terms to ensure ongoing protection of personal information.
In addition, even when HIPAA does not apply, according to the FTC, failing to take appropriate steps to keep consumers’ personal information secure constitutes unfair acts or practices in or affecting commerce in violation of Section 5(a) of the FTCA, 15 U.S.C § 45(a). The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities. Medical data is considered sensitive data that merits stronger safeguards. The FTC’s guidance for appropriately securing consumers’ personal information is similar to what is required by the HIPAA Security Rule.
Many foreign countries and governmental bodies, including the EU, Canada, Australia and other relevant jurisdictions, have laws and regulations concerning the collection and use of personal or sensitive data obtained from their residents or by businesses operating within their jurisdiction. For example, the European Commission recently adopted the General Data Protection Regulation, or the GDPR, effective on May 25, 2018, that supersedes current EU data protection legislation, imposes more stringent EU data protection requirements and provides for greater penalties for noncompliance. The GDPR applies to any company established in the EU as well as to those outside the EU if they collect and use personal data in connection with the offering goods or services to individuals in the EU or the monitoring of their behavior. The GDPR enhances data protection obligations for processors and controllers of personal data, including, for example, expanded disclosures about how personal information is to be used, limitations on retention of information, mandatory data breach notification requirements and onerous new obligations on services providers. Non-compliance with the GDPR can trigger steep fines of up to €20 million or 4% of total worldwide annual revenues, whichever is higher. Given the breadth and depth of changes in data protection obligations, meeting the GDPR’s requirements requires time, resources and a review of the technology and systems currently in use against the GDPR’s requirements.
We may be at risk of enforcement actions taken by certain EU data protection authorities while we continue to build our business practices to ensure that all transfers of personal data to us from the European Economic Area are conducted in compliance with all applicable regulatory obligations, the guidance of data protection authorities and evolving best practices. We may find it necessary to establish systems to maintain personal data originating from the EU in the European Economic Area, which may involve substantial expense and may cause us to need to divert resources from other aspects of our business, all of which may adversely affect our business.
Our failure to comply with applicable laws and regulations, or to protect such data, could result in enforcement actions against us, including fines, imprisonment of company officials and public censure, claims for damages by end-customers and other affected individuals, damage to our reputation and loss of goodwill, any of which could harm on our operations, financial performance, and business. Evolving and changing definitions of personal data and personal information, within the European Union, the United States, and elsewhere, may limit or inhibit our ability to operate or expand our business, including limiting strategic partnerships that may involve the sharing of data. Moreover, if the relevant laws and regulations change, or are interpreted and applied in a manner that is inconsistent with our data practices or the operation of our products, we may need to expend resources in order to change our business operations, data practices, or the manner in which our products operate. Even the perception of privacy concerns, whether or not valid, may harm our reputation and inhibit adoption of our products.
There is the risk that the limits we obtained for our cyber liability insurance may not cover the total loss experienced in the event of a data security incident, including the financial loss, legal costs, and business and reputational harm, particularly if there is an interruption to our systems. Additionally, there is the risk of a data privacy or security incident by an employee, which may expose us to liability. If personal information of our customers or employees is misappropriated, our reputation with our customers and employees may be injured resulting in loss of business and/or morale, and we may incur costs to remediate possible injury to our customers and employees or be required to pay fines or take other action with respect to judicial or regulatory actions arising out of such incidents.
48
Risks Related to Regulatory and Political Environment
The regulatory approval process is expensive, time consuming and uncertain, and may prevent us from obtaining approvals for the commercialization of Motiva Implants or our planned products.
The research, testing, manufacturing, labeling, approval, selling, import, export, marketing and distribution of medical devices are subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries, where regulations differ from country to country. Our products are registered to be sold in more than 80 countries, but we are not permitted to market our planned products in the United States until we receive the requisite approval or clearance from the FDA. We have not submitted an application or received marketing approval for Motiva Implants or any planned products in the United States. Obtaining PMA approval for sale of a medical device from the FDA can be a lengthy, expensive and uncertain process. In addition, failure to comply with FDA and other applicable U.S. and foreign regulatory requirements may subject us to administrative or judicially imposed sanctions, including the following:
•warning letters;
•civil or criminal penalties and fines;
•injunctions;
•suspension or withdrawal of regulatory approval;
•suspension of any ongoing clinical studies;
•voluntary or mandatory product recalls and publicity requirements;
•refusal to accept or approve applications for marketing approval of new devices or supplements to approved applications filed by us;
•restrictions on operations, including costly new manufacturing requirements; or
•seizure or detention of our products or import bans.
Prior to receiving approval to commercialize any of our planned products in the United States or abroad, we may be required to demonstrate with substantial evidence from preclinical and well-controlled clinical studies, and to the satisfaction of the FDA or other regulatory authorities abroad, that such planned products are safe and effective for their intended uses. Results from preclinical studies and clinical studies can be interpreted in different ways. Even if we believe the preclinical or clinical data for our planned products are promising, such data may not be sufficient to support approval by the FDA and other regulatory authorities. Administering any of our planned products to humans may produce undesirable side effects, which could interrupt, delay or cause suspension of clinical studies of our planned products and result in the FDA or other regulatory authorities denying approval of our planned products for any or all targeted indications.
Regulatory approval from the FDA is not guaranteed, and the approval process is expensive and may take several years. The FDA also has substantial discretion in the approval process. Despite the time and expense exerted, failure can occur at any stage, and we could encounter problems that cause us to abandon or repeat clinical studies, or perform additional preclinical studies and clinical studies. The number of preclinical studies and clinical studies that will be required for FDA approval varies depending on the planned product, the indication that the planned product is designed to address and the regulations applicable to any particular planned product. The FDA can delay, limit or deny approval of a planned product for many reasons, including, but not limited to, the following:
▪a planned product or one or more of its features may not be deemed safe or effective;
▪FDA officials may not find the data from preclinical studies and clinical studies sufficient;
▪the FDA might not approve our manufacturing or our third-party supplier’s processes or facilities; or
▪the FDA may change its approval policies or adopt new regulations.
If Motiva Implants or any planned products fail to demonstrate safety and efficacy in preclinical and clinical studies or do not gain regulatory approval, our business and results of operations will be harmed.
49
Even if we receive regulatory approval for a planned product, we will be subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense and subject us to penalties if we fail to comply with applicable regulatory requirements.
When a regulatory approval is obtained, the approved product and its manufacturer are subject to continual review by the FDA or non-U.S. regulatory authorities. Our regulatory approval for Motiva Implants, as well as any regulatory approval that we receive for Motiva Implants or for any planned products may be subject to limitations on the indicated uses for which the product may be marketed. Future approvals may contain requirements for potentially costly post-marketing follow-up studies to monitor the safety and efficacy of the approved product. In addition, we are subject to extensive and ongoing regulatory requirements by the FDA and other regulatory authorities with regard to the labeling, packaging, adverse event reporting, storage, advertising, promotion and recordkeeping for our products. In addition, we are required to comply with regulations regarding the manufacture of Motiva Implants, which include requirements related to quality control and quality assurance as well as the corresponding maintenance of records and documentation. Further, regulatory authorities must inspect these manufacturing facilities and determine they are in compliance with FDA good manufacturing practice requirements as set forth in the Quality System Regulation, or QSR, before the products can be approved. These facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with QSR regulations. If we or a third party discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory authority may impose restrictions on that product, the manufacturer or us, including requiring withdrawal of the product from the market or suspension of manufacturing.
We are subject to extensive and dynamic medical device regulation, which may impede or hinder the approval or sale of our products and, in some cases, may ultimately result in an inability to obtain approval of certain products or may result in the recall or seizure of previously approved products.
Our products, marketing, sales and development activities and manufacturing processes are subject to extensive and rigorous regulation by various regulatory agencies and governing bodies. Under the US Food, Drug and Cosmetic Act (FDC Act), medical devices must receive FDA clearance or approval or an exemption from such clearance or approval before they can be commercially marketed in the U.S. In the EU, we are required to comply with applicable medical device directives (including the Medical Devices Directive and the European Medical Device Regulation) and obtain CE Mark certification in order to market medical devices. In addition, exported devices are subject to the regulatory requirements of each country to which the device is exported. Many countries require that product approvals be renewed or recertified on a regular basis, generally every four to five years. The renewal or recertification process requires that we evaluate any device changes and any new regulations or standards relevant to the device and conduct appropriate testing to document continued compliance. Where renewal or recertification applications are required, they may need to be renewed and/or approved in order to continue selling our products in those countries. There can be no assurance that we will receive the required approvals for new products or modifications to existing products on a timely basis or that any approval will not be subsequently withdrawn or conditioned upon extensive post-market study requirements.
The European Union regulatory bodies finalized a new Medical Device Regulation (MDR) in 2017, which replaced the existing directives and provided three years for transition and compliance. After a one year delay, the MDR became effective on May 26, 2021. The MDR changes several aspects of the existing regulatory framework, such as updating clinical data requirements and introducing new ones, such as Unique Device Identification, or UDI. We and the Notified Bodies who will oversee compliance to the new MDR face uncertainties as the MDR is rolled out and enforced by the Commission and EEA Competent Authorities, creating risks in several areas, including the CE Marking process and data transparency, in the upcoming years.
Regulations regarding the development, manufacture and sale of medical devices are evolving and subject to future change. We cannot predict what impact, if any, those changes might have on our business. Failure to comply with regulatory requirements could have a material adverse effect on our business, financial condition and results of operations. Later discovery of previously unknown problems with a product or manufacturer could result in fines, delays or suspensions of regulatory clearances or approvals, seizures or recalls of products, physician advisories or other field actions, operating restrictions and/or criminal prosecution. We may also initiate field actions as a result of a failure to strictly comply with our internal quality policies. The failure to receive product approval clearance on a timely basis, suspensions of regulatory clearances, seizures or recalls of products, physician advisories or other field actions, or the withdrawal of product approval by regulatory authorities could have a material adverse effect on our business, financial condition or results of operations.
50
The medical technology industry is complex and intensely regulated at the federal, state, and local levels and government authorities may determine that we have failed to comply with applicable laws or regulations.
As a company which manufactures and distributes medical devices and technologies, we are subject to numerous regional, national and local laws and regulations. There are significant costs involved in complying with these laws and regulations. Moreover, if we are found to have violated any applicable laws or regulations, we could be subject to civil and/or criminal damages, fines, sanctions or penalties, including exclusion from participation in governmental healthcare programs. We may also be required to change our method of operations. These consequences could be the result of current conduct or even conduct that occurred a number of years ago. We also could incur significant costs merely if we become the subject of an investigation or legal proceeding alleging a violation of these laws and regulations. We cannot predict whether any government authority will determine that we are not operating in accordance with law, or whether the laws will change in the future and impact our business.
Under some circumstances, government investigations can also be initiated by private individuals under whistleblower provisions which may be incentivized by the possibility for private recoveries. Responding to inquiries and enforcement activities can be costly and disruptive to our business operations, even when the allegations are without merit. We also may be subject to other financial sanctions or be required to modify our operations. Any of these actions could have a material adverse effect on our business, financial condition and results of operations.
Health care reform measures could hinder or prevent our planned products’ commercial success.
In the United States, there have been, and we expect there will continue to be, a number of legislative and regulatory changes to the health care system in ways that could affect our future revenue and future profitability and the future revenue and future profitability of our potential customers. Federal and state lawmakers regularly propose and, at times, enact legislation that could result in significant changes to the health care system, some of which are intended to contain or reduce the costs of medical products and services. For example, one of the most significant health care reform measures in decades, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or PPACA, was enacted in 2010. The PPACA contains a number of provisions, including those governing enrollment in federal health care programs, reimbursement changes and fraud and abuse measures, all of which will impact existing government health care programs and will result in the development of new programs.
Some provisions of the PPACA have yet to be implemented, and there have been judicial and Congressional challenges to certain aspects of the PPACA, as well as prior efforts by the Trump administration to repeal or replace certain aspects of the PPACA. Furthermore, the Tax Cuts and Jobs Act passed in December of 2017 included a provision that would repeal one of the primary pillars of the law, the PPACA’s individual mandate penalty that essentially assessed a monetary penalty or fine on certain individuals who fail to maintain qualifying health coverage for all or part of a year. Congress may consider other legislation to repeal or replace elements of the PPACA on a provision-by-provision basis. We cannot assure you that the PPACA, as currently enacted or as amended in the future, will not adversely affect our business and financial results and we cannot predict how future federal or state legislative or administrative changes relating to health care reform will affect our business. Most recently, under President Biden, the Department of Justice dropped support of two Supreme Court cases challenging PPACA in addition to a case before the U.S. Court of Appeals for the Fifth Circuit. On January 28, 2021, President Biden signed an executive order to expand access to PPACA coverage, stating that it is the “policy” of the Biden administration to protect and strengthen the PPACA and directing agencies to consider suspending, revising, or rescinding actions related to President Trump’s executive orders that are inconsistent with this policy position. However, other legislators continue efforts to repeal and replace other elements of the PPACA. While the ultimate outcome of PPACA is not yet known, any changes that result in price controls reduce access to and reimbursement for care or add additional regulations may have an adverse effect on our financial condition and results of operations.
We cannot predict the impact that such actions against the PPACA or other health care reform under the Biden administration will have on our business, and there is uncertainty as to what healthcare programs and regulations may be implemented or changed at the federal and/or state level in the United States, or the effect of any future legislation or regulation. However, it is possible that such initiatives could have an adverse effect on our ability to obtain approval and/or successfully commercialize products in the United States in the future. For example, any changes that reduce, or impede the ability to obtain, reimbursement for the type of products we intend to
51
commercialize in the United States (or our products more specifically, if approved) or reduce medical procedure volumes could adversely affect our business plan to introduce our products in the United States.
In addition, other legislative changes have been proposed and adopted since the PPACA was enacted. For example, the Budget Control Act of 2011, among other things, created the Joint Select Committee on Deficit Reduction to recommend proposals for spending reductions to Congress. The Joint Select Committee did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, which triggered the legislation’s automatic reduction to several government programs, including aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013, which, due to subsequent legislative amendments to the statute, including the Bipartisan Budget Act of 2018, will remain in effect through 2027 unless additional Congressional action is taken. We cannot predict whether any additional legislative changes will affect our business.
There likely will continue to be legislative and regulatory proposals at the federal and state levels directed at containing or lowering the cost of health care. We cannot predict the initiatives that may be adopted in the future or their full impact. The continuing efforts of the government, insurance companies, managed care organizations and other payors of health care services to contain or reduce costs of health care may adversely affect:
▪our ability to set a price that we believe is fair for our products;
▪our ability to generate revenue and achieve or maintain profitability; and
▪the availability of capital.
Exposure to United Kingdom political developments, including the outcome of its withdrawal from membership in the European Union, could be costly and difficult to comply with and could seriously harm our business.
In June 2016, a referendum was held in the U.K. which resulted in a majority voting in favor of the U.K. withdrawing from the E.U. (commonly referred to as "Brexit"). Pursuant to legislation approved by the U.K. Parliament and the E.U. Parliament in January 2020, the U.K. withdrew from the E.U. with effect from 11 p.m. (GMT) on January 31, 2020 on the terms of a withdrawal agreement agreed between the U.K. and the E.U. in October 2019. On December 24, 2020, the U.K. and E.U. agreed to a trade deal, or the Trade and Cooperation Agreement, which was ratified by the U.K. on December 30, 2020 and the European Parliament and the Council of the European Union in April 2021, and became effective on May 1, 2021 (after being provisionally effective since January 1, 2021). There are still a number of areas of uncertainty in connection with the future of the U.K. and its relationship with the E.U. and the application and interpretation of the Trade and Cooperation Agreement, and Brexit related matters may take several years to be clarified and resolved. For example, because a significant proportion of the regulatory framework in the U.K. is currently derived from E.U. directives and regulations, Brexit could result in material changes to the regulatory regime applicable to many of our current operations. Although the Trade and Cooperation Agreement offers U.K. and E.U. companies preferential access to each other’s markets, ensuring imported goods will be free of tariffs and quotas, economic relations between the U.K. and the E.U. will now be on more restricted terms than existed previously. Therefore, at this time, we cannot predict the impact that the Trade and Cooperation Agreement and any future agreements contemplated under the terms of the Trade and Cooperation Agreement will have on our on our future business efforts to commercialize our products in the U.K. and E.U. Accordingly, it is possible that new terms of the Trade and Cooperation Agreement may adversely affect our operations and financial results. We are currently in the process of evaluating our own risks and uncertainties to ascertain what financial, trade, regulatory and legal implications the Trade and Cooperation Agreement could have on our operations in the U.K. and otherwise. Finally, uncertainty surrounding Brexit has contributed to recent fluctuations in the U.K. economy as a whole which could experience future disruptions. As a result, Brexit could cause financial and capital markets within and outside the U.K. or the E.U. to constrict, thereby negatively impacting our ability to finance our U. K. operations which could also have an adverse effect on our results of operations and financial condition.
These developments, or the perception that any of them could occur, may adversely affect European and worldwide economic and market conditions, significantly reduce global market liquidity and restrict the ability of key market participants to operate in certain financial markets and could contribute to instability in global financial and foreign exchange markets, including increased volatility in interest rates and foreign exchange rates. The potential impacts could adversely impact other global economies, and in particular, the European economy, a region which accounted for approximately 41% and 44% of our total revenues for the year ended December 31,
52
2021 and 2020, respectively. In the first quarter of 2019, we completed the migration of our CE Mark certificates, originally issued by BSI UK Notified Body, to BSI Group The Netherlands B.V., which is a European Notified Body designated in The Netherlands. We continue to actively monitor the ongoing potential impacts of Brexit and will seek to minimize its impact on our business through review of our existing regulatory requirements, contractual arrangements and obligations, particularly in the European region. Any of these effects of Brexit, among others, could adversely affect our business, business opportunities, results of operations, financial condition and cash flows.
Almost one year after it was introduced to the House of Commons, the Medical Devices Bill was granted Royal Assent on February 11, 2021, becoming law as the Medicines and Medical Devices Act 2021. The purpose of the Act is to create a structure for the UK Government to legislate for updates or amendments to the existing laws on human and veterinary medicine, clinical trials, and medical devices. We will need to comply with the Act on a going forward basis.
The political situation in the United States can affect the ability of our company to conduct business in certain areas or countries if new trade conditions are imposed or enforced by the U.S. government.
There could be negative consequences to our company’s revenue if the U.S. government unexpectedly changes its trade policies towards determined geographies or countries. These policy changes can include such things as trade barriers, which serve to limit or prevent international trade. The U.S. government may request additional funds or tariffs in exchange for the right to export items into the country. Tariffs or quotas may be used to protect domestic producers from foreign competition. Changes may include the modification or withdrawal of free trade agreements already in place. This also can have a large effect on the profits of our company because it either cuts revenues as a result of a tax on imports/exports or restricts the amount of revenues that can be earned.
Risks Related to Taxation
Tax authorities may disagree with our positions and conclusions regarding certain tax positions, resulting in unanticipated costs, taxes or non-realization of expected benefits.
A tax authority may disagree with tax positions that we have taken, which could result in increased tax liabilities. For example, the U.S. Internal Revenue Service or another tax authority could challenge the amounts paid between our affiliated companies pursuant to our intercompany arrangements and transfer pricing policies. A tax authority may take the position that material income tax liabilities, interest and penalties are payable by us, in which case, we expect that we might contest such assessment. Contesting such an assessment may be lengthy and costly and if we were unsuccessful in disputing the assessment, the implications could increase our anticipated effective tax rate, where applicable. In addition, we may be subject to additional tax liabilities, which could materially and adversely affect our business, financial condition and results of operations. The application, interpretation and enforcement value-added tax, or VAT, and other taxes and related regulations applicable to medical device companies is complex and evolving.
We are a multinational organization faced with increasingly complex tax issues in many jurisdictions, and changes in tax laws or their application to the operation of our business could adversely impact our operating results and our business.
We conduct operations in multiple jurisdictions, and we are subject to certain taxes, including income, sales and use, employment, value added and other taxes, in the United States and other jurisdictions in which we do business. A change in the tax laws in the jurisdictions in which we do business, including an increase in tax rates or an adverse change in the treatment of an item of income or expense, possibly with retroactive effect, could result in a material increase in the amount of taxes we incur.
Our determination of our tax liability is subject to review by applicable U.S. and foreign tax authorities. Any adverse outcome of such a review could harm our operating results and financial condition. The determination of our worldwide provision for income taxes and other tax liabilities requires significant judgment and, in the ordinary course of business, there are many transactions and calculations where the ultimate tax determination is complex and uncertain. Moreover, as a multinational business, we have subsidiaries that engage in many intercompany transactions in a variety of tax jurisdictions where the ultimate tax determination is complex and uncertain. The taxing authorities of the jurisdictions in which we operate may challenge our methodologies, which could impact our financial position and operating results. Historically, we have allocated some of our employees’ and contractors’ time across multiple business entities in the international jurisdictions in which we operate. If it were
53
determined that we had misclassified our employees’ or contractors’ employment status or certain of our expenditures under local laws, we may be subjected to penalties or be required to pay withholding taxes for, extend employee benefits to, provide compensation for unpaid overtime to, or otherwise incur substantially greater expenses with respect to such employees and contractors. Any of the foregoing circumstances could have a material adverse impact on our operating results and financial condition.
We are periodically reviewed and audited by tax authorities with respect to income and non-income taxes. Tax authorities may disagree with certain positions we have taken, and we may have exposure to additional income and non-income tax liabilities which could have an adverse effect on our operating results and financial condition. Such authorities could impose additional taxes, interest and penalties, claim that various withholding requirements apply to us or our subsidiaries or assert that benefits of tax treaties are not available to us or our subsidiaries. In addition, our future effective tax rates could be favorably or unfavorably affected by changes in tax rates, changes in the valuation of our deferred tax assets or liabilities, the effectiveness of our tax planning strategies, or changes in tax laws or their interpretation. Such changes could have an adverse impact on our financial condition.
As a result of these and other factors, the ultimate amount of tax obligations owed may differ from the amounts recorded in our financial statements and any such difference may harm our operating results in future periods in which we change our estimates of our tax obligations or in which the ultimate tax outcome is determined.
Our ability to use net operating losses to offset future taxable income and certain other tax attributes may be subject to certain limitations.
Federal and California laws impose restrictions on the utilization of net operating loss carryforwards and research and development credit carryforwards in the event of a change in ownership of the Company, which constitutes an “ownership change” as defined by Internal Revenue Code Sections 382 and 383. Generally, an ownership change occurs if the percentage of the value of the shares that are owned by one or more direct or indirect “five percent shareholders” increases by more than 50% over their lowest ownership percentage at any time during the applicable testing period. If we have experienced an “ownership change” at any time since our formation, we may already be subject to limitations on our ability to utilize our existing net operating losses and other tax attributes. We have not experienced an ownership change in the past that would materially impact the availability of its net operating losses and tax credits. Nevertheless, future changes in our share ownership, which may be outside of our control, may trigger an “ownership change” and, consequently, Section 382 and 383 limitations. We have not completed a Section 382 and 383 analysis to determine if an ownership change has occurred. Until such analysis is completed, we cannot be sure that the full amount of the existing net operating loss carryforwards will be available to us, even if we do generate taxable income before their expiration. In addition, under the newly enacted U.S. federal income tax law, federal net operating losses incurred in 2018 and in future years may be carried forward indefinitely, but the deductibility of such federal net operating losses is limited.
U.S. holders of our common shares may suffer adverse tax consequences if we are characterized as a passive foreign investment company.
A non-U.S. corporation will be classified as a passive foreign investment company, or PFIC, for U.S. federal income tax purposes, in any taxable year in which either (1) at least 75% of its gross income is passive income; or (2) at least 50% of the average quarterly value of its total gross assets is attributable to assets that produce “passive income” or are held for the production of passive income. Based on the project composition of our income and valuation of our assets, we do not believe we were a PFIC in 2020, and we do not expect to be a PFIC for our current taxable year or to become one in the future. However, because our PFIC status is subject to a number of uncertainties, neither we nor our tax advisors can provide any assurances regarding our PFIC status. If we are a PFIC for any taxable year during which a U.S. holder holds our common shares, the U.S. holder may be subject to adverse tax consequences. U.S. investors should consult their advisors regarding the application of these rules and the availability of any potential elections. See “Material British Virgin Island and U.S. Federal Income Tax Considerations.”
If a United States person is treated as owning at least 10% of our common shares, such holder may be subject to adverse U.S. federal income tax consequences.
If a United States person is treated as owning (directly, indirectly, or constructively) at least 10% of the value or voting power of our ordinary shares, such person may be treated as a “United States shareholder” with respect to each “controlled foreign corporation” in our group (if any). We may become a controlled foreign corporation. In addition, because our group includes one or more U.S. subsidiaries, certain of our non-U.S. subsidiaries could be
54
treated as controlled foreign corporations (regardless of whether or not we are treated as a controlled foreign corporation). A U.S. shareholder of a controlled foreign corporation may be required to report annually and include in its U.S. taxable income its pro rata share of “Subpart F income,” “global intangible low-taxed income,” and investments in U.S. property by controlled foreign corporations, regardless of whether we make any distributions. An individual that is a U.S. shareholder with respect to a controlled foreign corporation generally would not be allowed certain tax deductions or foreign tax credits that would be allowed to a U.S. shareholder that is a U.S. corporation. Failure to comply with these reporting obligations may subject a U.S. shareholder to significant monetary penalties and may prevent the statute of limitations with respect to such shareholder’s U.S. federal income tax return for the year for which reporting was due from starting. We cannot provide any assurances that we will assist investors in determining whether we or any of our non-U.S. subsidiaries is treated as a controlled foreign corporation or whether any investor is treated as a U.S. shareholder with respect to any such controlled foreign corporation or furnish to any U.S. shareholders information that may be necessary to comply with the aforementioned reporting and tax paying obligations. A U.S. investor should consult its advisors regarding the potential application of these rules to an investment in our common shares.
Discontinuation of preferential tax treatments we currently enjoy or other unfavorable changes in tax law could result in additional compliance obligations and costs.
Discontinuation of preferential tax treatments we currently enjoy or other unfavorable changes in tax law could result in additional compliance obligations and costs. We are currently the beneficiary of a tax holiday in Costa Rica pursuant to which we are subject to a tax at a 0% rate. However, there can be no assurance that we will continue to qualify for or receive such favorable tax treatment. If we fail to maintain such favorable tax treatment we may be subject to tax in Costa Rica at a significantly higher rate.
Risks Related to Ownership of Our Securities
Our share price may be volatile, and purchasers of our securities could incur substantial losses.
Our common shares have only recently become publicly traded, and we expect that the price of our common shares will likely be volatile. The securities markets in general, and the market for biotechnology and medical device companies in particular, have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. Additionally, the lack of an active market may impair the value of our common shares, or your ability to sell your shares at the time you wish to sell them or at a price that you consider reasonable. Although our common shares are listed on the Nasdaq Capital Market, if we fail to satisfy the continued listing standards, we could be de-listed, which would negatively impact the price of our common shares. The market price for our shares may be influenced by many factors, including the following:
•our ability to successfully commercialize, and realize revenues from sales of, Motiva Implants;
▪the success of competitive products or technologies;
▪results of clinical studies of Motiva Implants or planned products or those of our competitors;
•regulatory or legal developments in the United States and other countries, especially changes in laws or regulations applicable to our products;
•introductions and announcements of new products by us, our commercialization partners, or our competitors, and the timing of these introductions or announcements;
•actions taken by regulatory agencies with respect to our products, clinical studies, manufacturing processes or sales and marketing terms;
•variations in our financial results or those of companies that are perceived to be similar to us;
•the success of our efforts to acquire or in-license additional products or planned products;
•developments concerning our collaborations, including but not limited to those with our sources of manufacturing supply and our commercialization partners;
•developments concerning our ability to bring our manufacturing processes to scale in a cost-effective manner;
•announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments;
55
•developments or disputes concerning patents or other proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our products;
•our ability or inability to raise additional capital and the terms on which we raise it;
•the recruitment or departure of key personnel;
•changes in the structure of health care payment systems;
•negative shifts in the economy effecting the number of aesthetic breast procedures;
•market conditions in the pharmaceutical and biotechnology sectors;
•actual or anticipated changes in earnings estimates or changes in securities analyst recommendations regarding our common shares, other comparable companies or our industry generally;
•trading volume of our common shares;
•sales of our common shares by us or our shareholders;
•the impact of the COVID-19 pandemic;
•general economic, industry and market conditions; and
•the other risks described in this “Risk Factors” section.
These broad market and industry factors may harm the market price of our common shares, regardless of our operating performance. In the past, following periods of volatility in the market, securities class-action litigation has often been instituted against companies. Such litigation, if instituted against us, could result in substantial costs and diversion of management’s attention and resources, which could adversely affect our business, financial condition, results of operations and growth prospects.
We identified a material weakness in our internal control over financial reporting as of December 31, 2021 and 2020, and may identify additional material weaknesses in the future that may cause us to fail to meet our reporting obligations or result in material misstatements of our consolidated financial statements. If we fail to establish and maintain effective control over financial reporting, our ability to accurately and timely report our financial results could be adversely affected.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with U.S. generally accepted accounting principles. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. Prior to the completion of our IPO, we were a private company with limited accounting and compliance personnel and other resources to address our internal control over financial reporting.
It was determined that our primary user access controls (i.e. provisioning, de-provisioning, and quarterly user access review) to ensure appropriate segregation of duties that would adequately restrict user and privileged access to the financially relevant systems and data to appropriate Company personnel were not operating effectively. These user access control deficiencies resulted in a lack of segregation of duties with respect to certain user roles. Automated process-level controls and manual controls that are dependent upon the information derived from such financially relevant systems were also determined to be ineffective as a result of such deficiency. As a result, it was determined that a material weakness in our internal control over financial reporting existed as of December 31, 2021.
In addition, in connection with the preparation and audit of our 2020 financial statements, we had one material weakness. It was determined that we did not perform an adequate review over the manual consolidation process, resulting in an audit adjustment. We implemented a number of measures to address the material weakness, which was fully remediated as of December 31, 2021.
If additional material weaknesses or significant deficiencies in our internal control are discovered or occur in the future, it may materially adversely affect our ability to report our financial condition and results of operations in a timely and accurate manner and impact investor confidence in our Company.
56
The actions we have taken are subject to continued review, supported by confirmation and testing by management as well as audit committee oversight. The identification and remediation of additional material weaknesses in the future, could adversely affect our ability to report financial information, including our filing of quarterly or annual reports with the SEC on a timely and accurate basis and prohibit us from producing timely and accurate consolidated financial statements, which may adversely affect our share price and we may be unable to maintain compliance with Nasdaq listing requirements.
Our directors and principal shareholders continue to maintain the ability to control or significantly influence all matters submitted to shareholders for approval.
As of December 31, 2021, our executive officers, directors and shareholders who own more than 5% of our outstanding common shares, in the aggregate, assuming the exercise of all options held by such persons, beneficially owned shares representing approximately 56.6% of our common shares. As a result, if these shareholders were to choose to act together, they would be able to control or significantly influence all matters submitted to our shareholders for approval, as well as our management and affairs. For example, these shareholders, if they choose to act together, will control or significantly influence the election of directors and approval of any merger, consolidation or sale of all or substantially all of our assets. This concentration of voting power could delay or prevent an acquisition of our company on terms that other shareholders may desire.
Risks Related to Being a British Virgin Islands Company
Rights of shareholders under British Virgin Islands law differ from those under U.S. law, and, accordingly, you may have fewer protections as a shareholder.
Our corporate affairs are governed by our amended and restated memorandum and articles of association, the BVI Act, and the common law of the British Virgin Islands. The rights of shareholders to take legal action against our directors, actions by minority shareholders and the fiduciary responsibilities of our directors under British Virgin Islands law are to a large extent governed by the common law of the British Virgin Islands and by the BVI Act. The common law of the British Virgin Islands is derived in part from comparatively limited judicial precedent in the British Virgin Islands as well as from English common law, which has persuasive, but not binding, authority on a court in the British Virgin Islands. The rights of our shareholders and the fiduciary responsibilities of our directors under British Virgin Islands law are not as clearly established as they would be under statutes or judicial precedents in some jurisdictions in the United States. In particular, the British Virgin Islands has a less developed body of securities laws as compared to the United States, and some states (such as Delaware) have more fully developed and judicially interpreted bodies of corporate law. As a result of the foregoing, holders of our ordinary shares may have more difficulty in protecting their interests through actions against our management, directors or major shareholders than they would as shareholders of a U.S. company.
British Virgin Islands companies may not be able to initiate shareholder derivative actions, thereby depriving shareholders of one avenue to protect their interests.
British Virgin Islands companies may not have standing to initiate a shareholder derivative action in a federal court of the United States. The circumstances in which any such action may be brought, and the procedures and defenses that may be available in respect of any such action, may result in the rights of shareholders of a British Virgin Islands company being more limited than those of shareholders of a company organized in the United States. Accordingly, shareholders may have fewer alternatives available to them if they believe that corporate wrongdoing has occurred. The British Virgin Islands courts are also unlikely to recognize or enforce judgments of courts in the United States based on certain liability provisions of U.S. securities law, or to impose liabilities based on certain liability provisions of the U.S. securities laws that are penal in nature, in original actions brought in the British Virgin Islands. There is no statutory recognition in the British Virgin Islands of judgments obtained in the United States, although the courts of the British Virgin Islands will generally recognize and enforce the non-penal judgment of a non-U.S. court of competent jurisdiction without retrial on the merits. This means that even if shareholders were to sue us successfully, they may not be able to recover anything to make up for the losses suffered.
British Virgin Islands law differs from the laws in effect in the United States, and U.S. investors may have difficulty enforcing civil liabilities against us, our directors or members of senior management.
Under our amended and restated memorandum and articles of association, we may indemnify and hold our directors harmless against all claims and suits brought against them, subject to limited exceptions. Furthermore,
57
to the extent allowed by law, the rights and obligations among or between us, any of our current or former directors, officers and employees and any current or former shareholder will be governed exclusively by the laws of the British Virgin Islands and subject to the jurisdiction of the British Virgin Islands courts, unless those rights or obligations do not relate to or arise out of their capacities as such. Although there is doubt as to whether U.S. courts would enforce these provisions in an action brought in the United States, under U.S. securities laws, these provisions could make judgments obtained outside of the British Virgin Islands more difficult to enforce against our assets in the British Virgin Islands or jurisdictions that would apply British Virgin Islands law.
The laws of the British Virgin Islands provide limited protection for minority shareholders, so minority shareholders will have limited or no recourse if they are dissatisfied with the conduct of our affairs.
Under the laws of the British Virgin Islands, there is limited statutory law for the protection of minority shareholders other than the provisions of the BVI Act dealing with shareholder remedies, as summarized under “Description of Share Capital-Shareholders’ Rights Under British Virgin Islands Law Generally.” One protection under statutory law is that shareholders may bring an action to enforce the constituent documents of a British Virgin Islands company and are entitled to have the affairs of the Company conducted in accordance with the BVI Act and the amended and restated memorandum and articles of association of the Company. As such, if those who control the Company have disregarded the requirements of the BVI Act or the provisions of our amended and restated memorandum and articles of association, then the courts will likely grant relief. Generally, the areas in which the courts will intervene are the following: (i) an act complained of which is illegal; (ii) acts that constitute oppression, unfair discrimination or unfair prejudice against the minority where the wrongdoers control the Company; (iii) acts that infringe on the personal rights of the shareholders, such as the right to vote; and (iv) acts where we have not complied with provisions requiring approval of a special or extraordinary majority of shareholders, which are more limited than the rights afforded to minority shareholders under the laws of many states in the United States.
Provisions in our amended and restated memorandum and articles of association and under British Virgin Islands law could make an acquisition of us more difficult and may prevent attempts by our shareholders to replace or remove our current management.
Provisions in our amended and restated memorandum and articles of association may discourage, delay or prevent a merger, acquisition or other change in control of us that shareholders may consider favorable, including transactions in which shareholders might otherwise receive a premium for their shares. These provisions could also limit the price that investors might be willing to pay in the future for our common shares, thereby depressing the market price of our common shares. In addition, these provisions may frustrate or prevent any attempts by our shareholders to replace or remove our current management by making it more difficult for shareholders to replace members of our Board of Directors. Because our Board of Directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt by our shareholders to replace current members of our management team. Among others, these provisions include the following:
•our Board of Directors is divided into three classes with staggered three-year terms which may delay or prevent a change of our management or a change in control;
•our Board of Directors has the right to elect directors to fill a vacancy created by the expansion of our Board of Directors or the resignation, death or removal of a director, which will prevent shareholders from being able to fill vacancies on our Board of Directors;
•our shareholders are not able to act by written consent, and, as a result, a holder, or holders, controlling a majority of our shares are not able to take certain actions other than at annual shareholders’ meetings or special shareholders’ meetings;
•our amended and restated memorandum and articles of association do not allow cumulative voting in the election of directors, which limits the ability of minority shareholders to elect director candidates;
•amendments of our amended and restated memorandum and articles of association will require the approval of shareholders holding 66 2/3% of our outstanding voting shares (unless amended by the Board of Directors);
•our shareholders are required to provide advance notice and additional disclosures in order to nominate individuals for election to our Board of Directors or to propose matters that can be acted upon at a shareholders’ meeting, which may discourage or deter a potential acquiror from conducting a solicitation
58
of proxies to elect the acquiror’s own slate of directors or otherwise attempting to obtain control of our company; and
•our Board of Directors is able to issue, without shareholder approval, preferred shares with voting or other rights or preferences that could impede the success of any attempt to acquire us.
Moreover, because we are incorporated in the British Virgin Islands, we are governed by the provisions of BVI Business Companies Act, 2004, as amended, or the BVI Act, which provide for different shareholder rights than a Delaware corporation. See, for example, the risk factor titled “Rights of shareholders under British Virgin Islands law differ from those under U.S. law, and, accordingly, you may have fewer protections as a shareholder.”
59
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
Our principal executive offices are located in Alajuela, Costa Rica, where we occupy approximately 36,000 square feet of office, laboratory and manufacturing space. In order to increase our manufacturing capacity, we have constructed a new manufacturing facility of approximately 28,000 square feet, which began shipping manufactured product in March 2017. We exercised the option to purchase this manufacturing facility in June 2019.
We are in the process of expanding our manufacturing facilities and corporate offices in the Coyol Free Zone in Costa Rica. The project estimate includes approximately 170,000 square feet of facility space.
We also have office and warehouse space in Wommelgem, Belgium; Sao Paulo and Rio de Janiero, Brazil; Stockholm, Sweden; Barcelona, Spain; Rome, Italy; Miramar, Florida; London, England; Haar, Germany, Cavaillon, France and Buenos Aires, Argentina pursuant to a variety of leases that expire in 2022 through 2028.
ITEM 3. LEGAL PROCEEDINGS
We are not a party to any material legal proceeding required to be disclosed under Item 103 of Regulation S-K.
ITEM 4. MINE SAFETY DISCLOSURES
None.
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common shares have been traded on the Nasdaq Capital Market under the symbol “ESTA” since our initial public offering on July 23, 2018. Prior to this time, there was no public market for our common shares.
Holders
There were 24 shareholders of record of our common shares as of February 28, 2022. Certain shares are held in “street” name and, accordingly, the number of beneficial owners of such shares is not known or included in the foregoing number.
Sales of Unregistered Securities
None.
Dividends
We have not paid any cash dividends on our common shares since inception and do not anticipate paying cash dividends in the foreseeable future.
Purchases of Equity Securities by the Issuer or Affiliated Purchasers
There were no repurchases of shares of common shares made during the three months ended December 31, 2021.
ITEM 6. RESERVED
60
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with the consolidated financial statements and related notes that are included elsewhere in this Annual Report on Form 10-K. This discussion contains forward-looking statements based upon current expectations that involve risks and uncertainties. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of various factors, including those set forth under the sections contained in this Annual Report on Form 10-K entitled Item 1A. “Risk Factors” Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and Item 7A. “Quantitative and Qualitative Disclosures about Market Risk”. See “Special Note Regarding Forward-Looking Statements” preceding Part I of this Annual Report on Form 10-K.
Overview
Our line of silicone gel-filled breast implants, branded as Motiva Implants, is the centerpiece of our medical technology platform. Our post-market surveillance data (which was not generated in connection with a United States Food and Drug Administration, or FDA, pre-market approval, or PMA, study, but was patient or practitioner reported rather than collected at defined follow-ups) and published third-party data indicates that Motiva Implants show low rates of adverse events (including rupture, capsular contracture, and safety related reoperations) that we believe compare favorably with those of our competitors. We believe the proprietary technologies that differentiate our Motiva Implants enable improved safety and aesthetic outcomes and drive our revenue growth. We have developed other complementary products and services, which are aimed at further enhancing patient outcomes.
We have devoted a majority of our resources since inception to developing our Motiva Implants, which we began selling in October 2010. We have incurred net losses in each year since inception, and we have financed our operations primarily through equity financings and debt financings.
Our revenue for the years ended December 31, 2021 and 2020 was $126.7 million and $84.7 million, respectively, an increase of $42.0 million, or 49.6%. Net losses were $41.1 million for the year ended December 31, 2021 as compared to $38.1 million for the year ended December 31, 2020. As of December 31, 2021, we had an accumulated deficit of $206.4 million.
Our cash balance as of December 31, 2021 was $53.4 million.
We are in the process of expanding our manufacturing facilities and corporate offices in the Coyol Free Zone in Costa Rica. The initial $35.3 million project estimate includes approximately 145,000 square feet of facility space and would initially increase our manufacturing capacity by approximately 400,000 units per year, and potentially increase capacity by 800,000 units with an additional incremental $4.6 million investment in manufacturing equipment for a total facility space of approximately 170,000 square feet. We held the groundbreaking ceremony for our new Sulàyöm Innovation Campus in Costa Rica in the second quarter of 2021. Construction on the new building began following finalization and execution of certain contractual arrangements in the third quarter of 2021. The initial phase of construction of the cold-shell structure is being funded by the Coyol Free Zone, with Establishment Labs having the option to purchase the land and cold shell building. See Note 3, “Balance Sheet Accounts” for additional information regarding this construction project and our right to purchase the title to the land and cold shell building currently under construction.
We have made and continue to make significant investments in additional manufacturing capacity, marketing, customer service, and a direct sales force in certain territories like Brazil and several countries in Europe in order to drive and support further adoption of our Motiva Implants. We expect that we will continue to incur losses at least in the near term as we expand our organization to support planned sales growth, while also continuing to invest in research and development of our products, clinical trials to enable regulatory approval in the United States, and in other commercialization efforts. We also expect to incur significant additional expenditures as a public company. As a result, we may need to raise additional capital through equity and debt financings in order to fund our operations. Our operating results may fluctuate on a quarterly or annual basis in the future, and our growth or operating results may not be consistent with predictions made by securities analysts, if any. If we are unable to achieve our revenue growth objectives, we may not be able to achieve profitability.
61
Components of Results of Operations
Revenue
We commenced sales of our Motiva Implants in October 2010 and these products have historically accounted for the majority of our revenues. Sales of our Motiva breast implants accounted for over 98% of our revenues for the year ended December 31, 2021, and we expect our revenues to continue to be driven primarily by sales of these products. We primarily derive revenue from sales of our Motiva Implants to two types of customers: (1) medical distributors and (2) direct sales to physicians, hospitals, and clinics.
We recognize revenue related to the sales of products at the time of shipment, except for a portion of our direct sales revenue that is generated from the sale of consigned inventory maintained at physician, hospital, and clinic locations. For consignment sales, revenue is recognized at the time we are notified by the consignee that the product has been implanted. Our contracts with distributors do not typically contain right of return or price protection and have no post-delivery obligations.
We expect our revenue to increase as we enter new markets, expand awareness of our products in existing markets, and grow our distributor network and direct sales force. We also expect our revenue to fluctuate from quarter to quarter due to a variety of factors, including seasonal fluctuations in demand for Motiva Implants. We are also affected by foreign currency fluctuations.
Cost of Revenue and Gross Margin
Our implants are manufactured at our two facilities in Costa Rica. Cost of revenue is primarily the cost of silicone but also includes other raw materials, packaging, components, quality assurance, labor costs, as well as manufacturing and overhead expenses. Cost of revenue also includes depreciation expense for production equipment, and amortization of certain intangible assets.
We calculate gross margin as revenue less cost of revenue for a given period divided by revenue. Our gross margin may fluctuate from period to period depending, in part, on the efficiency and utilization of our manufacturing facilities, targeted pricing programs, and sales volume based on geography, customer and product type.
Operating Expenses
Sales, General and Administrative
Sales, general and administrative, or SG&A, expenses primarily consist of compensation, including salary, share-based compensation and employee benefits for our sales and marketing personnel, and for administrative personnel that support our general operations such as information technology, executive management, financial accounting, customer service, and human resources personnel. SG&A expenses also includes costs attributable to freight, marketing, sales support, travel, legal services, financial audit fees, insurance costs, and consulting services. We expect to incur additional SG&A expenses in connection with being a public company as we are no longer able to rely on the “emerging growth company” exemption we were previously afforded under the Jumpstart Our Business Startups Act of 2012, or JOBS Act.
We expect our SG&A expenses to continue to increase in absolute dollars for the foreseeable future as our business grows and we continue to invest in our sales, marketing, medical education, training and general administration resources to build our corporate infrastructure. However, we expect our SG&A expenses to decrease as a percentage of our revenue over the long term, although our SG&A expenses may fluctuate from period to period due to the timing of expenses related to our sales and marketing campaigns.
Research and Development
Our research and development, or R&D, activities primarily consist of engineering and research programs associated with our products under development, as well as R&D activities associated with our clinical development activities. Our R&D expenses primarily consist of compensation, including salary, share-based compensation and employee benefits for our R&D and clinical personnel. We also incur significant expenses for supplies, development prototypes, design and testing, clinical study costs and product regulatory and consulting expenses.
We expect our R&D expenses to continue to increase for the foreseeable future as we continue to advance our products under development, as well as initiate and prepare for additional clinical studies. We received an approval of an investigational device exemption, or IDE, from the FDA in March 2018 to initiate a clinical trial and enrolled the first patient in April 2018. In August 2019, we completed all patient surgeries for the IDE aesthetic
62
cohorts, which include primary augmentation and revision augmentation. As of June 2020, we successfully completed enrollment in the revision reconstruction sub-cohort and we are continuing to enroll subjects in the remaining reconstruction cohort. Although we continue to activate trial sites and secure Institutional Review Board approvals for our IDE clinical trial, the COVID-19 pandemic has delayed enrollment in the reconstruction cohort. We plan to complete enrollment of 800 patients in the study across 40 sites in the United States, Germany and Sweden in fiscal 2022. The results of the study are expected to support a PMA submission to the FDA. We estimate that total costs for this IDE clinical trial will be between $30.0 million and $40.0 million over ten years since the inception of the study. We also have other products under development for which we may be required to conduct clinical trials in future periods in order to receive regulatory approval to market these products.
Interest Expense
Interest expense consists primarily of cash and non-cash interest related to outstanding debt and amortization of debt discounts. As of December 31, 2021, we had $65.0 million in outstanding principal under our term loans.
Change in Fair Value of Derivative Instruments
Change in fair value of derivative instruments consists of changes in the fair value of the put and call option liabilities associated with outstanding debt instruments.
Change in Fair Value of Contingent Consideration
Change in fair value of contingent consideration consists of changes in the fair value of contingent equity consideration related to past asset acquisitions.
Other Income (Expense), Net
Other income (expense), net primarily consists of foreign currency gains/losses and interest income.
Income Tax Expense
Income tax expense consists primarily of income taxes in foreign jurisdictions in which we conduct business. Due to our history of losses, with the exception of Belgium and JAMM Technologies, Inc., we maintain a full valuation allowance for deferred tax assets including net operating loss carry-forwards, R&D tax credits, capitalized R&D and other book versus tax differences.
Business Update Regarding COVID-19
The COVID-19 pandemic has presented a substantial public health and economic challenge around the world, resulting in a disruption in our operations in fiscal 2020, and continued to adversely affect our business in fiscal 2021. Business uncertainties created by the COVID-19 pandemic have continued in fiscal 2022. We continue to closely monitor developments related to the COVID-19 pandemic and our decisions will continue to be driven by the health and well-being of our employees, our distributor and plastic surgeon customers, and their patients while maintaining operations to support our customers and their patients in the near-term.
•Surgery Deferrals: From late March 2020 to mid-May 2020, among other impacts on our business related to the pandemic, plastic surgeons and their patients deferred surgical procedures in which our products otherwise could have been used, including surgeries for our clinical trial participants. This decrease in demand for our products recovered to varying degrees in the latter half of May and into June 2020, though still below pre-pandemic levels through the rest of fiscal 2020, as certain geographies reopened after an initial improvement in COVID-19 infection rates and allowed plastic surgeons to resume providing procedures. However, the impact from the COVID-19 outbreak in fiscal 2020 has not had a material effect on the Company’s liquidity or financial position. During fiscal 2021, we saw an increase in surgical activity as women’s interest in plastic surgery exceeded pre-pandemic levels, and most geographies modified procedures to mitigate infection risk to allow returning to normalized operating levels. However, if a resurgence of infections is observed, we may see continued volatility through at least the duration of the pandemic as geographies respond to current local conditions. The duration of further deferrals of surgical procedures, the magnitude of such deferrals, the timing and extent of the economic impact of the pandemic, and the pace at which the economy recovers therefrom, cannot be determined at this time. We continue to work closely with our plastic surgeon customers, distributors and suppliers to navigate through this unforeseen event while maintaining flexible operations and investing for future growth.
•Operations: Our sales, marketing and research and development efforts have continued since the outbreak of the pandemic. However, the pandemic has adversely affected our business despite the steps taken to mitigate its impact. To protect the safety, health and well-being of our employees, distributor and plastic surgeon customers, and communities, we implemented preventative measures including travel
63
restrictions and requiring all office-based employees to work from home, except for those related to manufacturing and select others, as permitted under governmental orders.
Our manufacturing, distribution and supply chain has largely been uninterrupted, but could be disrupted as a result of the pandemic due to staffing shortages, production slowdowns, stoppages, or disruptions in delivery systems.
•2021 Results: The first and second quarters of 2021 resulted in record quarterly revenue, which pointed to the recovering global business environment and normalizing medical protocols as countries managed new COVID-19 infections. The third quarter of 2021 saw a decline in revenue as compared to the previous quarters in fiscal 2021, following more normalized seasonal patterns. Despite this decline, surgical activity remained above pre-pandemic levels during 2021, resulting in record annual revenue in fiscal 2021 and record quarterly revenue in the fourth quarter of 2021. The increase in revenue was driven by an increase in demand, especially in our European and Latin American markets, and our efforts to expand direct sales in multiple geographies.
•2020 Results: Given that the onset of COVID-19 occurred toward the end of the first quarter of 2020, our total revenue for the second quarter of 2020 was significantly lower compared to the same period in 2019. Our revenue for the third and fourth quarters of 2020, however, recovered and was comparable or exceeded the revenue in corresponding quarters of fiscal 2019.
•Outlook: At this time, the full extent of the impact of the COVID-19 pandemic on our business, financial condition and results of operations is uncertain and cannot be predicted with reasonable accuracy and will depend on future developments that are also uncertain and cannot be predicted with reasonable accuracy. However, management does not expect future results of operations to be materially impacted by the COVID-19 pandemic.
For additional information on the various risks posed by the COVID-19 pandemic on our business, financial condition and results of operations, please see Item 1A. Risk Factors in this report.
Consolidated Results of Operations
The following table sets forth our results of operations for the years presented, in dollars:
| 2021 | 2020 | ||||||||||
(in thousands) | |||||||||||
| Revenue | $ | 126,682 | $ | 84,676 | |||||||
| Cost of revenue | 41,278 | 32,174 | |||||||||
| Gross profit | 85,404 | 52,502 | |||||||||
| Operating expenses: | |||||||||||
| Sales, general and administrative | 92,229 | 66,625 | |||||||||
| Research and development | 18,315 | 13,793 | |||||||||
| Total operating expenses | 110,544 | 80,418 | |||||||||
| Loss from operations | (25,140) | (27,916) | |||||||||
| Interest expense | (9,062) | (9,373) | |||||||||
| Change in fair value of derivative instruments | 737 | 1,632 | |||||||||
| Change in fair value of contingent consideration | — | 304 | |||||||||
| Other income (expense), net | (6,247) | (2,664) | |||||||||
| Loss before income taxes | (39,712) | (38,017) | |||||||||
| Provision for income taxes | (1,427) | (104) | |||||||||
| Net loss | $ | (41,139) | $ | (38,121) | |||||||
64
Comparison of the Year Ended December 31, 2021 and 2020
| 2021 | 2020 | ||||||||||
(in thousands) | |||||||||||
Revenue | $ | 126,682 | $ | 84,676 | |||||||
| Cost of revenue | 41,278 | 32,174 | |||||||||
| Gross profit | $ | 85,404 | $ | 52,502 | |||||||
Gross margin | 67.4 | % | 62.0 | % | |||||||
Revenue
Revenue increased $42.0 million, or 49.6%, to $126.7 million for the year ended December 31, 2021, as compared to $84.7 million for the year ended December 31, 2020. The increase was primarily due to share gains in global markets and the global economies recovering from the COVID-19 pandemic declared in March 2020 that resulted in multiple geographies experiencing restrictions on elective surgical procedures and a shut-down or slow down in business activity deemed to be non-essential.
Cost of Revenue and Gross Margin
Cost of revenue increased $9.1 million, or 28.3%, to $41.3 million for the year ended December 31, 2021, compared to $32.2 million for the year ended December 31, 2020. The increase in cost of revenue is in line with the increase in revenue due to the economic recovery from the COVID-19 pandemic.
Gross margin increased to 67.4% for the year ended December 31, 2021, compared to 62.0% for the year ended December 31, 2020, primarily due to the benefit of geographic mix, greater operating efficiencies, and enhanced manufacturing planning capabilities.
Operating Expenses
| 2021 | 2020 | ||||||||||
| (in thousands) | |||||||||||
| Operating expenses: | |||||||||||
| Sales, general and administrative | $ | 92,229 | $ | 66,625 | |||||||
| Research and development | 18,315 | 13,793 | |||||||||
| Total operating expenses | $ | 110,544 | $ | 80,418 | |||||||
Sales, General and Administrative Expense
SG&A expense increased $25.6 million, or 38.4%, to $92.2 million for the year ended December 31, 2021, compared to $66.6 million for the year ended December 31, 2020. The increase in SG&A expense was primarily due to a $12.1 million increase in personnel and related costs due to increased headcount, a $5.6 million increase in sales and marketing expenses, a $5.0 million increase in consulting fees in part due to added costs in preparation for compliance with Section 404(b) of the Sarbanes-Oxley Act, and a $1.9 million increase in freight as we were impacted by increased shipping volumes as well as rate increases globally.
Research and Development Expense
R&D expense increased $4.5 million, or 32.8%, to $18.3 million for the year ended December 31, 2021, compared to $13.8 million for the year ended December 31, 2020. The increase in R&D expense was primarily due to a $4.8 million increase in personnel cost due to increased headcount, partially offset by a $0.6 million decrease in expenditures related to our IDE clinical trial in the United States, primarily consisting of fees to third parties to manage the clinical trial.
Interest Expense
Interest expense decreased $0.3 million, or 3.3%, to $9.1 million for the year ended December 31, 2021, as
65
compared to $9.4 million for the year ended December 31, 2020. The decrease was primarily due to $0.7 million of direct costs to amend the Madryn Credit Agreement in August 2020.
Change in Fair Value of Derivative Instruments
Change in fair value of derivative instruments for the year ended December 31, 2021 resulted in a gain of $0.7 million, as compared to a gain of $1.6 million for the year ended December 31, 2020. The change in fair value of derivative instruments was due to changes in the fair value of Madryn derivatives embedded in the Madryn Credit Agreement we originally entered into in August 2017.
Change in Fair Value of Contingent Consideration
The liability related to the contingent consideration was extinguished in 2020 when the remaining contingently-issuable shares were issued.
Provision for Income Taxes
Provision for income taxes increased $1.3 million to $1.4 million for the year ended December 31, 2021, compared to $0.1 million for the year ended December 31, 2020. The change in the provision for income taxes is primarily due to certain true-ups in foreign jurisdictions partially offset by the release of the valuation allowance for JAMM Technologies, Inc.
Other Income (Expense), Net
Other income (expense), net increased $3.6 million to $6.3 million for the year ended December 31, 2021, compared to $2.7 million for the year ended December 31, 2020. The increase was primarily due to the foreign currency fluctuations of Brazilian real and the euro as compared to the U.S. dollar in fiscal 2020 and 2021, resulting in a foreign currency transaction loss of $5.6 million, the majority of which remains unrealized, for the year ended December 31, 2021, compared to $1.7 million for the year ended December 31, 2020.
Comparison of the Year Ended December 31, 2020 and 2019
The discussion related to our results of operations and changes in financial condition for 2020 compared to 2019 is incorporated by reference to Part II, Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations in our Annual Report on Form 10-K for the year ended December 31, 2020, which was filed with the SEC on March 15, 2021.
Liquidity and Capital Resources
As of December 31, 2021, we had an accumulated deficit of $206.4 million. Since our inception, we have generated losses and expect to continue to generate losses in the near term. We have financed our operations through a combination of equity financings and debt financings, and from cash generated from operations, primarily from the collection of accounts receivable resulting from sales. Our historical cash outflows have primarily been associated with cash used for operating activities such as expansion of our sales and marketing and distributor infrastructure, investing in inventory, R&D activities, asset acquisitions, capital improvements and other working capital needs. As of December 31, 2021 and 2020, we had cash of $53.4 million and $84.5 million, respectively.
Our short term liquidity requirements consist primarily of operating expenses and interest payments on the Madryn Credit Agreement. We believe that our available cash and cash from operations will be sufficient to satisfy our liquidity requirements for at least the next 12 months, including our contractual and other obligations summarized below under “Material Cash Requirements” section. Our long-term liquidity needs consist primarily of operating expenses, including expected increases in SG&A and R&D expenses related to our IDE clinical trial and product development and funds necessary to pay for the interest and principal payment on the Madryn Credit Agreement. Our liquidity assumptions may prove to be incorrect, and we could utilize our available financial resources sooner than we currently expect.
Our future capital requirements will depend on many factors, including:
▪the degree and rate of market adoption of our products;
▪the cost and timing of our regulatory activities, especially the IDE clinical trial to obtain regulatory approval for our Motiva Implants in the United States;
▪the emergence of new competing technologies and products;
▪the costs of R&D activities we undertake to develop and expand our products;
▪the costs of commercialization activities, including sales, marketing and manufacturing;
66
▪the level of working capital required to support our growth; and
▪our need for additional personnel, information technology or other operating infrastructure to support our growth and operations as a public company.
We may need to raise additional capital to execute our business plan. If we are unable to raise additional capital when desired, or on terms acceptable to us, our business, results of operations, and financial condition would be adversely affected.
Cash Flows
The discussion related to our cash flows for 2019 is incorporated by reference to Part II, Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations in our Annual Report on Form 10-K for the year ended December 31, 2020, which was filed with the SEC on March 15, 2021.
The following table sets forth the primary sources and uses of cash for each of the years presented below:
| 2021 | 2020 | ||||||||||
| (in thousands) | |||||||||||
| Net cash provided by (used in): | |||||||||||
| Operating activities | $ | (27,532) | $ | (12,510) | |||||||
| Investing activities | (7,163) | (5,559) | |||||||||
| Financing activities | 4,052 | 64,670 | |||||||||
| Effect of exchange rate changes on cash | (465) | 267 | |||||||||
| Net (decrease) increase in cash | $ | (31,108) | $ | 46,868 | |||||||
Net Cash Used in Operating Activities
Net cash used in operating activities of $27.5 million for the year ended December 31, 2021 was primarily comprised of a net loss of $41.1 million and a $0.7 million change in fair value of financial instruments, partially offset by $10.4 million of share-based compensation expense, $3.7 million of non-cash depreciation expense, $2.1 million of non-cash interest expense due to accretion of debt discounts, $4.2 million unrealized foreign currency loss, as well as changes in operating assets and liabilities of $6.4 million.
Net cash used in operating activities of $12.5 million for the year ended December 31, 2020 was comprised of a net loss of $38.1 million and a $1.6 million change in fair value of financial instruments, partially offset by $5.7 million of share-based compensation expense, $3.3 million of non-cash depreciation expense, $1.7 million of non-cash interest expense due to accretion of debt discounts, $2.4 million unrealized foreign currency loss, a $1.2 million change in provision for inventory obsolescence, and a $0.4 million change in provision for doubtful accounts, as well as changes in operating assets and liabilities of $12.6 million.
Net Cash Used in Investing Activities
Net cash used in investing activities of $7.2 million for the year ended December 31, 2021 primarily consisted of $2.4 million in purchases of property and equipment, $0.4 million of cash paid for past asset acquisitions, $1.4 million of purchases of intangibles and $2.9 million of cash paid for capital expenditures on construction in progress.
Net cash used in investing activities of $5.6 million for the year ended December 31, 2020 primarily consisted of $2.1 million in purchases of property and equipment, $1.7 million of cash paid for past asset acquisitions, $1.5 million of purchases of intangibles and $0.3 million of cash paid for capital expenditures on construction in progress.
Net Cash Provided by Financing Activities
Net cash provided by financing activities of $4.1 million for the year ended December 31, 2021 primarily consisted of $4.6 million in proceeds received for stock option exercises, which were partially offset by $0.2 million in repayment on finance leases and a $0.4 million tax payment related to shares withheld upon vesting of restricted stock.
67
Net cash provided by financing activities of $64.7 million for the year ended December 31, 2020 primarily reflected $63.9 million in proceeds received from the issuance of common shares in the follow-on public offering, net of underwriters’ discount and issuance costs, $1.2 million in proceeds received for stock option exercises, which were partially offset by $0.3 million in repayment on finance leases and a $0.2 million tax payment related to shares withheld upon vesting of restricted stock.
Material Cash Requirements
The following table provides a summary of our material cash requirements from known contractual and other obligations, including commitments for capital expenditures, as of December 31, 2021:
| (in thousands) | 2022 | 2023 | 2024 | 2025 | 2026 | Thereafter | Total | |||||||||||||||||||||||||||||||||||||
Debt obligations - principal (1) | $ | — | $ | — | $ | — | $ | 65,000 | $ | — | $ | — | $ | 65,000 | ||||||||||||||||||||||||||||||
Debt obligations - Interest payments (1) | 6,901 | 6,901 | 6,920 | 5,157 | — | — | 25,879 | |||||||||||||||||||||||||||||||||||||
Future minimum lease payments (2) | 563 | 535 | 504 | 429 | 404 | 436 | 2,871 | |||||||||||||||||||||||||||||||||||||
Consideration payable related to asset acquisition (3) | 546 | — | — | — | — | — | 546 | |||||||||||||||||||||||||||||||||||||
License and software commitments (4) | 1,229 | 1,163 | 319 | 33 | — | — | 2,744 | |||||||||||||||||||||||||||||||||||||
| $ | 9,239 | $ | 8,599 | $ | 7,743 | $ | 70,619 | $ | 404 | $ | 436 | $ | 97,040 | |||||||||||||||||||||||||||||||
(1) Contractual obligations related to the Madryn Credit Agreement. The interest payment were projected using the constant LIBOR rate as of December 31, 2021. See below under “Indebtedness” and Note 6 “Debt” for additional details. | ||||||||||||||||||||||||||||||||||||||||||||
(2) Contractual obligations related to the minimum lease payments and interest on our operating leases. See Note 7 “Leases” for additional details. | ||||||||||||||||||||||||||||||||||||||||||||
(3) The last installment and milestone payment, earned in fiscal 2021, related to an asset purchase agreement entered in October 2018. | ||||||||||||||||||||||||||||||||||||||||||||
(4) Contractual obligations related to our current contracts for software solutions and support. | ||||||||||||||||||||||||||||||||||||||||||||
In August 2021, we entered into a contract with the Zona Franca Coyol, S.A., or CFZ, to begin construction of a new manufacturing facility in Costa Rica. The costs for improvement of the land and construction of a cold shell building are being paid for by CFZ and, upon completion, we will have the option to purchase the title to the land and cold shell building for approximately $12.6 million or to lease the facility at a to be determined price. Subject to purchase of the land and cold shell building, we will have the option to buy an adjacent lot of land for approximately $2.8 million and engage CFZ to construct an additional manufacturing facility.
Indebtedness
Madryn Debt
On August 24, 2017, we entered into a credit agreement, or the Madryn Credit Agreement, with Madryn Health Partners, LP, or Madryn, as administrative agent, and a syndicate of lenders that matures September 30, 2025.
The Madryn Credit Agreement, as amended, provides for term loans in a maximum aggregate principal amount of $65.0 million, all of which is outstanding as of December 31, 2021.
Borrowings under the Madryn Credit Agreement bear interest at a rate equal to 3-month LIBOR plus 8.0% per annum provided that no default has occurred. In an event of a default, the interest would increase by an additional 4.0% per annum. The effective interest rate under the amended Madryn Credit Agreement is 18.4%, and the weighted average interest rate was approximately 10.6% at December 31, 2021. No principal payments are due on the term loans until the final maturity date on September 30, 2025.
We also determined that the Madryn Credit Agreement contained put options, which are mandatory repayment provisions related to liquidity events or an event of default and a call option related to voluntary repayment option. We revalue the embedded derivatives as of each reporting period and record the change in the fair value in the consolidated statement of operations as other income or expense (see Note 5).
See Note 6 “Debt” for additional information regarding the Madryn Credit Agreement.
68
Critical Accounting Policies, Significant Judgments and Use of Estimates
This discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with the generally accepted accounting principles in the United States of America, or GAAP. The preparation of these consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of assets and liabilities, revenue and expenses, and the disclosure of contingent assets and liabilities. Our estimates are based on our historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities. Actual results may differ from these estimates. We believe that the critical accounting policies discussed below are essential to understanding our historical and future performance, as these policies relate to the more significant areas involving management’s estimates and judgments.
The full extent to which the COVID-19 pandemic will directly or indirectly impact our business, results of operations and financial condition, including sales, operational expenses, manufacturing, research and development costs, IDE clinical trial enrollment and related costs, and employee-related compensation, will depend on future developments that are highly uncertain, including as a result of new information that may emerge concerning COVID-19 and the actions taken to contain it or treat COVID-19, as well as the economic impact on local, regional, national and international customers and markets. We have made estimates of the impact of COVID-19 within our financial statements and there may be changes to those estimates in future periods. Actual results may differ from these estimates.
Revenue Recognition
The Company recognizes revenue related to sales of products to distributors or directly to customers in markets where it has regulatory approval, net of discounts and allowances. The Company recognizes revenue in accordance with Accounting Standards Codification, or ASC, 606, Revenue from Contracts with Customers. ASC 606 requires the Company to recognize revenue to depict the transfer of goods or services to a customer at an amount that reflects the consideration it expects to receive in exchange for those goods or services.
The Company recognizes revenue related to the sales of products to distributors at the time of shipment of the product, which represents the point in time when the distributor has taken ownership and assumed the risk of loss and the required revenue recognition criteria are satisfied. The Company’s distributors are obligated to pay within specified terms regardless of when, or if, they sell the products. The Company’s contracts with distributors typically do not contain right of return or price protection and have no post-delivery obligations.
The Company recognizes revenue when title to the product and risk of loss transfer to customers, provided there are no remaining performance obligations required of the Company or any written matters requiring customer acceptance. The Company allows for the return of product from direct customers in certain regions in limited instances within fifteen days after the original sale and records estimated sales returns as a reduction of sales in the same period revenue is recognized. Appropriate reserves are established for anticipated sales returns based on historical experience, recent gross sales and any notification of pending returns. Actual sales returns in any future period are inherently uncertain and thus may differ from the estimates. If actual sales returns differ significantly from the estimates, an adjustment to revenue in the current or subsequent period is recorded. As of December 31, 2021 and 2020, an allowance of $10,000 and $54,000 was recorded for product returns, respectively.
A portion of the Company’s revenue is generated from the sale of consigned inventory maintained at physician, hospital, and clinic locations. For these products, revenue is recognized at the time the Company is notified by the consignee that the product has been implanted, not when the consigned products are delivered to the consignee’s warehouse.
The Company has a limited warranty for the shelf life of breast implants, which is five years from the time of manufacture. Estimated warranty obligations are recorded at the time of sale. The Company also offers a warranty to patients in the event of rupture and a replacement program for capsular contracture events, provided certain registration requirements are met. Revenue for extended warranties is recognized ratably over the term of the agreement. To date, these warranty and program costs have been de minimis. The Company will continue to evaluate the warranty reserve policies for adequacy considering claims history.
Deferred revenue primarily consists of payments received in advance of meeting revenue recognition criteria. The Company has received payments from distributors to provide distribution exclusivity within a geographic area and recognizes deferred revenue on a ratable basis over the term of such contractual distribution relationship.
69
Additionally, the Company has received payments from customers in direct markets prior to surgical implantation and recognizes deferred revenue at the time the Company is notified by the customer that the product has been implanted. For all arrangements, any revenue that has been deferred and is expected to be recognized beyond one year is classified as long-term deferred revenue and included in “Other liabilities, long-term” on the consolidated balance sheets.
Research and Development
Costs related to research and development, or R&D, activities are expensed as incurred. R&D costs primarily include personnel costs, materials, clinical expenses, regulatory expenses, product development, consulting services, and outside research activities, all of which are directly related to research and development activities.
The Company estimates IDE clinical trial expenses based on the services performed, pursuant to contracts with research institutions and clinical research organizations that conduct and manage clinical trials on its behalf. In accruing service fees, the Company estimates the time period over which services will be performed and the level of patient enrollment and activity expended in each period. If the actual timing of the performance of services or the level of effort varies from the estimate, the Company will adjust the accrual accordingly.
Accounts Receivable and Allowance for Doubtful Accounts
Accounts receivable is stated at invoice value less estimated allowances for returns and doubtful accounts. The Company continually monitors customer payments and maintains an allowance for estimated losses resulting from customers’ inability to make required payments. In evaluating the Company’s ability to collect outstanding receivable balances, the Company considers various factors including the age of the balance, the creditworthiness of the customer, which is assessed based on ongoing credit evaluations and payment history, and the customer’s current financial condition. In cases where there are circumstances that may impair a specific customer’s ability to meet its financial obligations, an allowance is recorded against amounts due, which reduces the net recognized receivable to the amount reasonably believed to be collectible.
Inventory and Cost of Revenue
Inventory is stated at the lower of cost to purchase or manufacture the inventory or the net realizable value of such inventory. Cost is determined using the standard cost method which approximates actual costs using the first-in, first-out basis. The Company regularly reviews inventory quantities considering actual losses, projected future demand, and remaining shelf life to record a provision for excess and slow-moving inventory.
Long-Lived Assets
The Company reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset might not be recoverable. When such an event occurs, management determines whether there has been impairment by comparing the anticipated undiscounted future net cash flows to the related asset group’s carrying value. If an asset is considered impaired, the asset is written down to fair value, which is determined based either on discounted cash flows or appraised value, depending on the nature of the asset. There were no impairment charges, or changes in estimated useful lives recorded during the years ended December 31, 2021 and 2020.
Debt and Embedded Derivatives
The Company applies the accounting standards for derivatives and hedging and for distinguishing liabilities from equity when accounting for hybrid contracts. The Company accounts for convertible debt instruments when the Company has determined that the embedded conversion options should not be bifurcated from their host instruments in accordance with ASC 470-20 Debt with Conversion and Other Options (see Note 6).
The Company uses option pricing valuation models to determine the fair value of embedded derivatives and records any change in fair value as a component of other income or expense in the consolidated statements of operations (see Note 5).
Income Taxes
The Company records income taxes using the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in the Company’s consolidated financial statements or income tax returns. In estimating future tax consequences, expected future events, enactments or changes in the tax law or rates are considered. Valuation allowances are provided when necessary to reduce deferred tax assets to the amount expected to be realized.
70
The Company operates in various tax jurisdictions and is subject to audit by various tax authorities.
The Company records uncertain tax positions based on a two-step process whereby (1) a determination is made as to whether it is more likely than not that the tax positions will be sustained based on the technical merits of the position and (2) for those tax positions that meet the more-likely-than-not recognition threshold the Company recognizes the largest amount of tax benefit that is greater than 50% likely to be realized upon ultimate settlement with the related tax authority. The Company’s policy is to recognize interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense. Significant judgment is required in the identification of uncertain tax positions and in the estimation of penalties and interest on uncertain tax positions.
There were no material uncertain tax positions as of December 31, 2021 and 2020.
Foreign Currency
The financial statements of the Company’s foreign subsidiaries whose functional currencies are the local currencies are translated into U.S. dollars for consolidation as follows: assets and liabilities at the exchange rate as of the balance sheet date, stockholders’ equity at the historical rates of exchange, and income and expense amounts at the average exchange rate for the period. Translation adjustments resulting from the translation of the subsidiaries’ accounts are included in “Accumulated other comprehensive income” as equity in the consolidated balance sheet. Transactions denominated in currencies other than the applicable functional currency are converted to the functional currency at the exchange rate on the transaction date. At period end, monetary assets and liabilities are remeasured to the functional currency using exchange rates in effect at the balance sheet date. Non-monetary assets and liabilities are remeasured at historical exchange rates. Gains and losses resulting from foreign currency transactions are included within “Other income (expense), net” in the consolidated statement of operations. For the year ended December 31, 2021, foreign currency transaction loss amounted to $5.6 million as compared to a foreign currency transaction loss of $1.7 million for the year ended December 31, 2020.
Share-Based Compensation
The Company measures and recognizes compensation expense for all share-based awards in accordance with the provisions of ASC 718, Stock Compensation. Share-based awards granted include stock options, restricted stock units, or RSUs, and restricted stock awards, or RSAs. Share-based compensation expense for stock options and RSAs granted to employees is measured at the grant date based on the fair value of the awards and is recognized as an expense ratably on a straight-line basis over the requisite service period. The fair value of options to purchase shares granted to employees is estimated on the grant date using the Black-Scholes option valuation model.
The calculation of share-based compensation expense requires the Company to make assumptions and judgments about the variables used in the Black-Scholes model, including the expected term, expected volatility of the underlying common shares, risk-free interest rate and dividends.
See Note 10 “Share-Based Compensation” for additional information.
Recent Accounting Pronouncements
Please refer to Note 2 - “Summary of Significant Accounting Policies” in the notes to the consolidated financial statements included in this Form 10-K for information on recent accounting pronouncements and the expected impact on our unaudited consolidated financial statements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate Risk
We had cash of $53.4 million and $84.5 million as of December 31, 2021 and 2020, respectively. We manage our cash portfolio for operating and working capital purposes. Our cash balances are held in bank checking accounts, and we believe that we do not have any material exposure to changes in the fair value of our cash portfolio as a result of changes in interest rates.
Foreign Currency Exchange Risk
To date, the majority of our revenue has been denominated in U.S. dollars, Brazilian reals and euros. Some of our operating expenses are subject to fluctuations due to changes in foreign currency exchange rates, particularly changes in the euro and Brazilian real. Fluctuations in foreign currency exchange rates may cause us to recognize transaction gains and losses in our consolidated statements of operations. For the year ended
71
December 31, 2021, foreign currency transaction loss amounted to $5.6 million primarily related to the remeasurement of transactions denominated in the U.S. dollar into the Brazilian real and the euro as part of the financial reporting consolidation process under GAAP. The Brazilian real has been experiencing a weakening since December 31, 2017 as compared to the U.S. dollar with a slight recovery experienced in the second quarter of fiscal year 2021. We have not engaged in any foreign currency hedging activities. As our international operations grow, we will continue to reassess our approach to managing the risks relating to fluctuations in foreign currency exchange rates. During the year ended December 31, 2021, the effect of an immediate 10% adverse change in foreign exchange rates on foreign-denominated accounts as of December 31, 2021 would have had an impact of approximately 10.0% on revenues and would have impacted our net loss by a commensurate amount.
Inflation Risk
We do not believe that inflation had a significant impact on our results of operations for any periods presented in our consolidated financial statements.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The financial statements required to be filed pursuant to this Item 8 are appended to this report beginning on page F‑1. An index of those financial statements is included in Part IV, Item 15 below.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
As of December 31, 2021, the end of the period covered by this Annual Report on Form 10-K, our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. Disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act) are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is (i) recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and (ii) accumulated and communicated to the Company’s management, including its principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure. Based on the evaluation of our disclosure controls and procedures, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were not effective as of the end of the period covered by this annual report at the reasonable assurance level.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rule 13a-15(f) of the Exchange Act. Our management has assessed the effectiveness of our internal control over financial reporting as of December 31, 2021 using the criteria established in “Internal Control—Integrated Framework” (2013), issued by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the Company’s annual or interim financial statements will not be prevented or detected on a timely basis. Based on that assessment and due to the material weakness described below, our management has concluded that the Company’s internal control over financial reporting was not effective as of December 31, 2021. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the Company’s annual or interim financial statements will not be prevented or detected on a timely basis.
Material Weaknesses in Internal Control and Plan for Remediation
It was determined that our primary user access controls (i.e. provisioning, de-provisioning, and quarterly user access review) to ensure appropriate segregation of duties that would adequately restrict user and privileged access to the financially relevant systems and data to appropriate Company personnel were not operating effectively. These user access control deficiencies resulted in a lack of segregation of duties with respect to
72
certain user roles. Automated process-level controls and manual controls that are dependent upon the information derived from such financially relevant systems were also determined to be ineffective as a result of such deficiency. As a result, it was determined that a material weakness in our internal control over financial reporting existed as of December 31, 2021.
Our management believes the material weakness identified above has not had any material effect on our financial results. However, we are currently reviewing our controls and procedures related to this material weakness and expect to implement changes prior to the end of fiscal 2022, including identifying any specific areas within our information technology systems that require additional resources to mitigate this material weakness.
Remediation of Previously Reported Material Weakness in Internal Control over Financial Reporting
We disclosed in our Annual Report on Form 10-K for the year ended December 31, 2020, that the following material weakness in our internal control over financial reporting existed as of December 31, 2020: a lack of adequate review over the manual consolidation process, resulting in an audit adjustment.
We implemented a number of measures to address the material weakness identified as of December 31, 2020. We improved policies and procedures and designed and documented more effective controls that addressed the relevant risks in order to remediate the previously identified material weakness in addition to engaging a third-party consulting firm to assist us with the continuing implementation of SAP, which is a global information technology solution designed to address, among other items, the elements which gave rise to the material weakness.
During the fourth quarter of fiscal 2021, we completed the necessary testing to conclude that the material weakness identified as of December 31, 2020 has been fully remediated as of December 31, 2021.
Changes in Internal Control over Financial Reporting
There was no change in our internal control over financial reporting that occurred during the three months ended December 31, 2021 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting. Although we have altered some work routines due to the COVID-19 pandemic, the changes in our work environment, including remote work arrangements, have not materially impacted our internal control over financial reporting and have not adversely affected the Company’s ability to maintain operations.
Limitations on Effectiveness of Controls and Procedures
Our disclosure controls and procedures and internal control over financial reporting are designed to provide reasonable assurance of achieving the desired control objectives. Our management recognizes that any control system, no matter how well designed and operated, is based upon certain judgments and assumptions and cannot provide absolute assurance that its objectives will be met. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints and that management is required to apply judgment in evaluating the benefits of possible controls and procedures relative to their costs. Similarly, an evaluation of controls cannot provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud, if any, have been detected.
Attestation Report of the Independent Registered Public Accounting Firm
The effectiveness of our internal control over financial reporting as of December 31, 2021 has been audited by Marcum LLP, an independent registered public accounting firm, as stated in their report which is included below.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
ON INTERNAL CONTROL OVER FINANCIAL REPORTING
To the Shareholders and Board of Directors of
Establishment Labs Holdings Inc.
Adverse Opinion on Internal Control over Financial Reporting
We have audited Establishment Labs Holdings Inc.’s (the "Company") internal control over financial reporting as of December 31, 2021, based on criteria established in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. In our opinion, because of the effect of the material weakness described below on the achievement of the objectives of the control criteria, the Company
73
has not maintained effective internal control over financial reporting as of December 31, 2021, based on criteria established in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
A material weakness is a control deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the Company's annual or interim financial statements will not be prevented or detected on a timely basis. The following material weakness have been identified and included in “Management's Annual Report on Internal Control Over Financial Reporting”:
The Company’s primary user access controls (i.e. provisioning, de-provisioning, and quarterly user access review) to ensure appropriate segregation of duties that would adequately restrict user and privileged access to the financially relevant systems and data to appropriate Company personnel were not operating effectively. These user access control deficiencies resulted in a lack of segregation of duties with respect to certain user roles. Automated process-level controls and manual controls that are dependent upon the information derived from such financially relevant systems were also determined to be ineffective as a result of such deficiency.
This material weakness was considered in determining the nature, timing and extent of audit tests applied in our audit of the fiscal 2021 consolidated financial statements, and this report does not affect our report dated March 1, 2022 on those financial statements.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the consolidated balance sheets as of December 31, 2021 and the related consolidated statements of operations, comprehensive loss, shareholders’ equity, and cash flows for the year ended December 31, 2021 of the Company and our report dated March 1, 2022 expressed an unqualified opinion on those financial statements.
Basis for Opinion
The Company's management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying "Management Annual Report on Internal Control Over Financial Reporting". Our responsibility is to express an opinion on the Company's internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of the inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that degree of compliance with the policies or procedures may deteriorate.
/s/ Marcum LLP
Marcum LLP
74
Costa Mesa, California
March 1, 2022
ITEM 9B. OTHER INFORMATION
None.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Code of Business Conduct and Ethics
Our Board has adopted a Code of Business Conduct and Ethics that applies to all of our employees, officers and directors, including our chief executive officer, and other executive and senior financial officers. A copy of the code is posted under "Corporate Governance" in the Investors section of our website at https://establishmentlabs.com. If we make any substantive amendments to, or grant any waivers from, the Code of Business Conduct and Ethics for any officer or director, we will disclose the nature of such amendment or waiver on our website or in a current report on Form 8-K.
The remaining information required by this Item is incorporated by reference from the information in our Proxy Statement for our 2022 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
ITEM 11. EXECUTIVE COMPENSATION
Incorporated by reference from the information in our Proxy Statement for our 2022 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
Incorporated by reference from the information in our Proxy Statement for our 2022 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Incorporated by reference from the information in our Proxy Statement for our 2022 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
Incorporated by reference from the information in our Proxy Statement for our 2022 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
PART IV
ITEM 15. EXHIBIT AND FINANCIAL STATEMENT SCHEDULES
The following statements are filed as part of this Annual Report on Form 10-K:
1. Financial Statements.
A listing of the Consolidated Financial Statements, related notes and Report of Independent Registered Public Accounting is set forth on page F-1 in this Annual Report on Form 10-K.
75
2. Financial Statement Schedules.
All schedules have been omitted since the required information is not present or is not present in amounts sufficient to require submission of a schedule, or because the information required is included in the financial statements or related notes.
3. Index to Exhibits.
| Exhibit Number | Description of Exhibit | Incorporation by Reference | ||||||||||||
| 3.1 | Incorporated by reference from Registrant’s Form S-1/A filed July 9, 2018. | |||||||||||||
| 4.1 | Incorporated by reference from Registrant’s Form S-1/A filed July 13, 2018. | |||||||||||||
| 4.2 | Incorporated by reference from Registrant’s Form S-1 filed June 21, 2018. | |||||||||||||
| 4.3 | Incorporated by reference from Registrant’s Form S-1 filed June 21, 2018. | |||||||||||||
| 4.4 | Incorporated by reference from Registrant’s Form S-1 filed June 21, 2018. | |||||||||||||
| 4.5 | Incorporated by reference from Registrant’s Form S-1 filed June 21, 2018. | |||||||||||||
| 4.6 | Incorporated by reference from Registrant’s Annual Report on Form 10-K filed March 16, 2019. | |||||||||||||
| 10.1 | Incorporated by reference from Registrant’s Current Report on Form 8-K filed December 10, 2021. | |||||||||||||
| 10.2+ | Incorporated by reference from Registrant’s Form S-1 filed June 21, 2018. | |||||||||||||
| 10.3+ | Incorporated by reference from Registrant’s Form S-1/A filed July 9, 2018. | |||||||||||||
| 10.4+ | Incorporated by reference from Registrant’s Form S-1/A filed July 9, 2018. | |||||||||||||
| 10.5 | Incorporated by reference from Registrant’s Form S-1 filed June 21, 2018. | |||||||||||||
| 10.6 | Incorporated by reference from Registrant’s Form S-1 filed June 21, 2018. | |||||||||||||
| 10.7‡ | Incorporated by reference from Registrant’s Form S-1/A filed July 13, 2018. | |||||||||||||
10.8‡ | Incorporated by reference from Registrant’s Form S-1/A filed July 13, 2018. | |||||||||||||
| 10.9 | Incorporated by reference from Registrant’s Form S-1/A filed July 13, 2018. | |||||||||||||
| 10.10 | Incorporated by reference from Registrant’s Form S-1/A filed July 13, 2018. | |||||||||||||
| 10.11 | Incorporated by reference from Registrant’s Form S-1/A filed July 13, 2018. | |||||||||||||
| Exhibit Number | Description of Exhibit | Incorporation by Reference | ||||||||||||
| 10.12 | Incorporated by reference from Registrant’s Form S-1 filed June 21, 2018. | |||||||||||||
10.13‡ | Incorporated by reference from Registrant’s Form S-1 filed July 13, 2018. | |||||||||||||
| 10.14 | Incorporated by reference from Registrant’s Current Report on Form 8-K filed October 10, 2018. | |||||||||||||
| 10.15+ | Incorporated by reference from Registrant’s Current Report on Form 8-K filed December 28, 2018. | |||||||||||||
| 10.16+ | Incorporated by reference from Registrant’s Current Report on Form 8-K filed December 28, 2018. | |||||||||||||
| 10.17 | Incorporated by reference from Registrant’s Current Report on Form 8-K filed June 18, 2019. | |||||||||||||
| 10.18 | Incorporated by reference from Registrant’s Current Report on Form 8-K filed June 26, 2019. | |||||||||||||
10.19‡ | Incorporated by reference from Registrant’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2019. | |||||||||||||
| 10.20 | Incorporated by reference from Registrant’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2020. | |||||||||||||
10.21‡ | Incorporated by reference from Registrant’s Current Report on Form 8-K filed September 25, 2020. | |||||||||||||
| 10.22+ | Incorporated by reference from Registrant’s Current Report on Form 8-K filed May 18, 2021. | |||||||||||||
10.23 | Filed herewith. | |||||||||||||
| 10.24+ | Filed herewith. | |||||||||||||
| 10.25+ | Filed herewith. | |||||||||||||
| 10.26+ | Filed herewith. | |||||||||||||
| 21.1 | Filed herewith. | |||||||||||||
| 23.1 | Filed herewith. | |||||||||||||
| 31.1 | Filed herewith. | |||||||||||||
| 31.2 | Filed herewith. | |||||||||||||
| 32.1* | Furnished herewith. | |||||||||||||
| Exhibit Number | Description of Exhibit | Incorporation by Reference | ||||||||||||
| 101.INS | Inline XBRL Instance Document - the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document | |||||||||||||
| 101.SCH | XBRL Taxonomy Extension Schema Document | |||||||||||||
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | |||||||||||||
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | |||||||||||||
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document | |||||||||||||
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document | |||||||||||||
| 104 | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101) | |||||||||||||
+ Indicates management contract or compensatory plan or arrangement.
‡ Portions omitted, or to be omitted, pursuant to a request for confidential treatment.
* The certifications furnished in Exhibit 32.1 hereto is deemed to accompany this Annual Report on Form 10-K and will not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, except to the extent that the registrant specifically incorporates it by reference.
ITEM 16. FORM 10-K SUMMARY
None.
| PAGE | |||||
Report of Independent Registered Public Accounting Firm | F-1 | ||||
| Consolidated Financial Statements | |||||
F-2 | |||||
F-3 | |||||
F-4 | |||||
F-5 | |||||
F-6 | |||||
F-8 | |||||
79
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors of
Establishment Labs Holdings Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Establishment Labs Holdings Inc. (the “Company”) as of December 31, 2021 and 2020, the related consolidated statements of operations, comprehensive loss, shareholders’ equity and cash flows for each of the three years in the period ended December 31, 2021, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2021, in conformity with accounting principles generally accepted in the United States of America.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) ("PCAOB"), the Company's internal control over financial reporting as of December 31, 2021, based on the criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in 2013 and our report dated March 1, 2022, expressed an adverse opinion on the effectiveness of the Company’s internal control over financial reporting because of the existence of a material weakness.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
Critical audit matters are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. We determined that there are no critical audit matters.
/s/ Marcum LLP
Marcum LLP (688)
We have served as the Company’s auditor since 2016.
Costa Mesa, CA
March 1, 2022
F-1
ESTABLISHMENT LABS HOLDINGS INC.
Consolidated Balance Sheets
(In thousands, except share data)
| December 31, | |||||||||||
| 2021 | 2020 | ||||||||||
| Assets | |||||||||||
| Current assets: | |||||||||||
| Cash | $ | 53,415 | $ | 84,523 | |||||||
Accounts receivable, net of allowance for doubtful accounts of $1,221 and $1,143 | 24,437 | 19,127 | |||||||||
| Inventory, net | 28,407 | 23,210 | |||||||||
| Prepaid expenses and other current assets | 7,012 | 5,439 | |||||||||
| Total current assets | 113,271 | 132,299 | |||||||||
| Long-term assets: | |||||||||||
| Property and equipment, net of accumulated depreciation | 18,658 | 16,202 | |||||||||
| Goodwill | 465 | 465 | |||||||||
| Intangible assets, net of accumulated amortization | 4,371 | 4,148 | |||||||||
| Right-of-use operating lease assets, net | 2,206 | 2,610 | |||||||||
| Other non-current assets | 558 | 664 | |||||||||
| Total assets | $ | 139,529 | $ | 156,388 | |||||||
| Liabilities and shareholders’ equity | |||||||||||
| Current liabilities: | |||||||||||
| Accounts payable | $ | 14,475 | $ | 9,722 | |||||||
| Accrued liabilities | 16,236 | 14,532 | |||||||||
| Other liabilities, short-term | 1,178 | 1,646 | |||||||||
| Total current liabilities | 31,889 | 25,900 | |||||||||
| Long-term liabilities: | |||||||||||
| Note payable, Madryn, net of debt discount and issuance costs | 51,906 | 49,832 | |||||||||
| Madryn put option | 703 | 1,440 | |||||||||
| Operating lease liabilities, non-current | 1,900 | 1,923 | |||||||||
| Other liabilities, long-term | 2,392 | 2,332 | |||||||||
| Total liabilities | 88,790 | 81,427 | |||||||||
| Commitments and contingencies (Note 15) | |||||||||||
| Shareholders’ equity: | |||||||||||
Common shares - zero par value, unlimited amount of shares authorized at December 31, 2021 and 2020; 24,488,335 and 23,925,789 shares issued at December 31, 2021 and 2020, respectively; 24,080,265 and 23,517,719 shares outstanding at December 31, 2021 and 2020, respectively | 219,737 | 213,471 | |||||||||
| Additional paid-in-capital | 36,584 | 26,717 | |||||||||
Treasury shares, at cost, 408,070 shares held at December 31, 2021 and 2020 | (2,854) | (2,854) | |||||||||
| Accumulated deficit | (206,385) | (165,246) | |||||||||
| Accumulated other comprehensive income | 3,657 | 2,873 | |||||||||
| Total shareholders’ equity | 50,739 | 74,961 | |||||||||
| Total liabilities and shareholders’ equity | $ | 139,529 | $ | 156,388 | |||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-2
ESTABLISHMENT LABS HOLDINGS INC.
Consolidated Statements of Operations
(In thousands, except share and per share data)
| Year Ended December 31, | ||||||||||||||||||||
| 2021 | 2020 | 2019 | ||||||||||||||||||
| Revenue | $ | 126,682 | $ | 84,676 | $ | 89,565 | ||||||||||||||
| Cost of revenue | 41,278 | 32,174 | 34,704 | |||||||||||||||||
| Gross profit | 85,404 | 52,502 | 54,861 | |||||||||||||||||
| Operating expenses: | ||||||||||||||||||||
| Sales, general and administrative | 92,229 | 66,625 | 70,811 | |||||||||||||||||
| Research and development | 18,315 | 13,793 | 14,991 | |||||||||||||||||
| Total operating expenses | 110,544 | 80,418 | 85,802 | |||||||||||||||||
| Loss from operations | (25,140) | (27,916) | (30,941) | |||||||||||||||||
| Interest income | 23 | 15 | 4 | |||||||||||||||||
| Interest expense | (9,062) | (9,373) | (8,696) | |||||||||||||||||
| Change in fair value of derivative instruments | 737 | 1,632 | 3,052 | |||||||||||||||||
| Change in fair value of contingent consideration | — | 304 | 276 | |||||||||||||||||
| Other income (expense), net | (6,270) | (2,679) | (1,205) | |||||||||||||||||
| Loss before income taxes | (39,712) | (38,017) | (37,510) | |||||||||||||||||
| Provision for income taxes | (1,427) | (104) | (640) | |||||||||||||||||
| Net loss | $ | (41,139) | $ | (38,121) | $ | (38,150) | ||||||||||||||
| Basic and diluted net loss per share | $ | (1.72) | $ | (1.63) | $ | (1.86) | ||||||||||||||
| Weighted average outstanding shares used for basic and diluted net loss per share | 23,972,722 | 23,316,102 | 20,541,528 | |||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
ESTABLISHMENT LABS HOLDINGS INC.
Consolidated Statements of Comprehensive Loss
(In thousands)
| Year Ended December 31, | |||||||||||||||||
| 2021 | 2020 | 2019 | |||||||||||||||
| Net loss | $ | (41,139) | $ | (38,121) | $ | (38,150) | |||||||||||
| Other comprehensive income: | |||||||||||||||||
| Foreign currency translation gain | 784 | 2,182 | 242 | ||||||||||||||
| Other comprehensive gain | 784 | 2,182 | 242 | ||||||||||||||
| Comprehensive loss | $ | (40,355) | $ | (35,939) | $ | (37,908) | |||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
ESTABLISHMENT LABS HOLDINGS INC.
Consolidated Statements of Shareholders’ Equity
(In thousands, except share data)
| Common Shares | Treasury Shares | Additional Paid-In Capital | Accumulated Deficit | Accumulated Other Comprehensive Income | Total | ||||||||||||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Balance at January 1, 2019 | 20,672,025 | $ | 145,709 | (408,070) | $ | (2,854) | $ | 15,156 | $ | (88,975) | $ | 449 | $ | 69,485 | |||||||||||||||||||||||||||||||||||||||
| Issuance of common shares in partial settlement of contingent consideration | 33,333 | 630 | — | — | — | — | — | 630 | |||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common shares in an asset acquisition | 12,404 | 337 | — | — | — | — | — | 337 | |||||||||||||||||||||||||||||||||||||||||||||
| Warrant exercises | 87,321 | 129 | — | — | (73) | — | — | 56 | |||||||||||||||||||||||||||||||||||||||||||||
| Stock option exercises | 80,991 | 712 | — | — | — | — | — | 712 | |||||||||||||||||||||||||||||||||||||||||||||
| Share-based compensation | 181,516 | 182 | — | — | 6,344 | — | — | 6,526 | |||||||||||||||||||||||||||||||||||||||||||||
| Shares withheld to cover income tax obligation upon vesting of restricted stock | (10,550) | (11) | — | — | (213) | — | — | (224) | |||||||||||||||||||||||||||||||||||||||||||||
| Foreign currency translation gain | — | — | — | — | — | — | 242 | 242 | |||||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | (38,150) | — | (38,150) | |||||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2019 | 21,057,040 | 147,688 | (408,070) | (2,854) | 21,214 | (127,125) | 691 | 39,614 | |||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock, net of underwriters’ discount and issuance costs | 2,628,571 | 63,855 | — | — | — | — | — | 63,855 | |||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common shares in settlement of contingent consideration | 33,334 | 618 | — | — | — | — | — | 618 | |||||||||||||||||||||||||||||||||||||||||||||
| Stock option exercises | 143,402 | 1,246 | — | — | — | — | — | 1,246 | |||||||||||||||||||||||||||||||||||||||||||||
| Share-based compensation | 70,910 | 71 | — | — | 5,650 | — | — | 5,721 | |||||||||||||||||||||||||||||||||||||||||||||
| Shares withheld to cover income tax obligation upon vesting of restricted stock | (7,468) | (7) | — | — | (147) | — | — | (154) | |||||||||||||||||||||||||||||||||||||||||||||
| Foreign currency translation gain | — | — | — | — | — | — | 2,182 | 2,182 | |||||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | (38,121) | — | (38,121) | |||||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2020 | 23,925,789 | 213,471 | (408,070) | (2,854) | 26,717 | (165,246) | 2,873 | 74,961 | |||||||||||||||||||||||||||||||||||||||||||||
| Stock option exercises | 521,316 | 6,226 | — | — | (141) | — | — | 6,085 | |||||||||||||||||||||||||||||||||||||||||||||
| Shares withheld to cover strike price upon cashless option exercise | (1,879) | (3) | (3) | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Share-based compensation | 48,124 | 48 | — | — | 10,359 | — | — | 10,407 | |||||||||||||||||||||||||||||||||||||||||||||
| Shares withheld to cover income tax obligation upon vesting of restricted stock | (5,015) | (5) | — | — | (351) | — | — | (356) | |||||||||||||||||||||||||||||||||||||||||||||
| Foreign currency translation gain | — | — | — | — | — | — | 784 | 784 | |||||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | (41,139) | — | (41,139) | |||||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2021 | 24,488,335 | $ | 219,737 | (408,070) | $ | (2,854) | $ | 36,584 | $ | (206,385) | $ | 3,657 | $ | 50,739 | |||||||||||||||||||||||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
ESTABLISHMENT LABS HOLDINGS INC.
Consolidated Statements of Cash Flows
(In thousands)
| Year Ended December 31, | ||||||||||||||||||||
| 2021 | 2020 | 2019 | ||||||||||||||||||
| Cash flows from operating activities: | ||||||||||||||||||||
| Net loss | $ | (41,139) | $ | (38,121) | $ | (38,150) | ||||||||||||||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||||||||||
| Depreciation and amortization | 3,718 | 3,348 | 3,288 | |||||||||||||||||
| Provision for doubtful accounts | 200 | 375 | 111 | |||||||||||||||||
| Provision for inventory obsolescence | 338 | 1,180 | 143 | |||||||||||||||||
| Provision for deferred income taxes | 7 | (274) | — | |||||||||||||||||
| Share-based compensation | 10,407 | 5,721 | 6,526 | |||||||||||||||||
| Loss from disposal of property and equipment | 170 | 170 | 67 | |||||||||||||||||
| Unrealized foreign currency loss, net | 4,200 | 2,386 | 3,533 | |||||||||||||||||
| Amortization of right-to-use asset | 406 | 363 | — | |||||||||||||||||
| Gain from write-off of liability | (736) | — | — | |||||||||||||||||
| Change in fair value of derivative instruments | (737) | (1,632) | (3,052) | |||||||||||||||||
| Change in fair value of contingent consideration | — | (304) | (276) | |||||||||||||||||
| Amortization of debt discount | 2,074 | 1,690 | 2,428 | |||||||||||||||||
| Changes in operating assets and liabilities: | ||||||||||||||||||||
| Accounts receivable | (6,695) | 3,805 | (5,511) | |||||||||||||||||
| Inventory | (7,644) | 4,786 | (3,372) | |||||||||||||||||
| Prepaid expenses and other current assets | (1,611) | 1,130 | (2,538) | |||||||||||||||||
| Other assets | 90 | (277) | (54) | |||||||||||||||||
| Accounts payable | 4,959 | (636) | 1,830 | |||||||||||||||||
| Accrued liabilities | 4,726 | 3,108 | 4,644 | |||||||||||||||||
| Operating lease liabilities | (408) | (318) | — | |||||||||||||||||
| Other liabilities | 143 | 990 | 400 | |||||||||||||||||
| Net cash used in operating activities | (27,532) | (12,510) | (29,983) | |||||||||||||||||
| Cash flows from investing activities: | ||||||||||||||||||||
| Purchases of property and equipment | (2,425) | (2,106) | (6,288) | |||||||||||||||||
| Cash used in asset acquisitions | (434) | (1,652) | (767) | |||||||||||||||||
| Cost incurred for intangible assets | (1,447) | (1,484) | (711) | |||||||||||||||||
| Capital expenditures on construction in progress | (2,857) | (317) | — | |||||||||||||||||
| Net cash used in investing activities | (7,163) | (5,559) | (7,766) | |||||||||||||||||
| Cash flows from financing activities: | ||||||||||||||||||||
| Borrowings under Madryn credit agreement, net of issuance costs | — | — | 24,748 | |||||||||||||||||
| Repayments on finance leases | (175) | (277) | (242) | |||||||||||||||||
| Proceeds from issuance of common shares, net of underwriters’ discount and issuance costs | — | 63,855 | — | |||||||||||||||||
| Cash used to repurchase warrants | — | — | (2,261) | |||||||||||||||||
| Proceeds from stock option exercises | 4,583 | 1,246 | 712 | |||||||||||||||||
| Proceeds from warrant exercises | — | — | 56 | |||||||||||||||||
| Tax payments related to shares withheld upon vesting of restricted stock | (356) | (154) | (224) | |||||||||||||||||
| Net cash provided by financing activities | 4,052 | 64,670 | 22,789 | |||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
ESTABLISHMENT LABS HOLDINGS INC.
Consolidated Statements of Cash Flows
(In thousands)
| Year Ended December 31, | ||||||||||||||||||||
| 2021 | 2020 | 2019 | ||||||||||||||||||
| Effect of exchange rate changes on cash | (465) | 267 | (24) | |||||||||||||||||
| Net (decrease)/increase in cash | (31,108) | 46,868 | (14,984) | |||||||||||||||||
| Cash at beginning of period | 84,523 | 37,655 | 52,639 | |||||||||||||||||
| Cash at end of period | $ | 53,415 | $ | 84,523 | $ | 37,655 | ||||||||||||||
| Supplemental disclosures: | ||||||||||||||||||||
| Cash paid for interest | $ | 6,927 | $ | 6,962 | $ | 5,947 | ||||||||||||||
| Cash paid for income taxes | $ | 652 | $ | 316 | $ | 649 | ||||||||||||||
| Supplemental disclosures of non-cash investing and financing activities: | ||||||||||||||||||||
| Unpaid balance for property and equipment | $ | 22 | $ | 210 | $ | 465 | ||||||||||||||
| Assets acquired under finance leases | $ | — | $ | — | $ | 69 | ||||||||||||||
| Equity consideration in an asset acquisition | $ | — | $ | — | $ | 337 | ||||||||||||||
| Consideration payable related to asset acquisition | $ | 546 | $ | 858 | $ | 1,271 | ||||||||||||||
| Inventory acquired in an asset acquisition | $ | — | $ | 1,009 | $ | 1,257 | ||||||||||||||
| Issuance of common shares in settlement of contingent consideration | $ | — | $ | 618 | $ | 630 | ||||||||||||||
| Intangible assets acquired in an asset acquisition | $ | — | $ | 138 | $ | — | ||||||||||||||
| Cashless option exercises | $ | 1,640 | $ | — | $ | — | ||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-7
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
1. Formation and Business of the Company
Formation and Business of the Company
Establishment Labs Holdings Inc. and its wholly owned subsidiaries (collectively “the Company”) is a global company that manufactures and markets innovative medical devices for aesthetic and reconstructive plastic surgery. The Company was established in the British Virgin Islands on October 9, 2013, at which time Establishment Labs, S.A., the Costa Rican manufacturing company, was reincorporated as a wholly-owned subsidiary. As of December 31, 2021, the Company also has wholly-owned subsidiaries in the United States (JAMM Technologies, Inc. and Motiva USA LLC), Brazil (Establishment Labs Produtos para Saude Ltda), Belgium (European Distribution Center Motiva BVBA), France (Motiva Implants France SAS), Sweden (Motiva Nordica AB), Switzerland (JEN-Vault AG), the United Kingdom (Motiva Implants UK Limited), Italy (Motiva Italy S.R.L), Spain (Motiva Implants Spain, S.L.), Austria (Motiva Austria GmbH), Germany (Motiva Germany GmbH) and Argentina (Motiva Argentina S.R.L). Substantially all of the Company’s revenues are derived from the sale of silicone gel-filled breast implants, branded as Motiva Implants.
The main manufacturing activities are conducted at two manufacturing facilities in Costa Rica. In 2010, the Company began operating under the Costa Rica free zone regime (Régimen de Zona Franca), which provides for reduced income tax and other tax obligations pursuant to an agreement with the Costa Rican authorities.
The Company’s products are approved for sale in Europe, the Middle East, Latin America, and Asia. The Company sells its products internationally through a combination of distributors and direct sales to customers.
The Company is pursuing regulatory approval to commercialize its products in the United States. The Company received approval for an investigational device exemption, or IDE, from the United States Food and Drug Administration, or FDA, in March 2018 to initiate a clinical trial in the United States for its Motiva Implants. In August 2019, the Company completed all patient surgeries for the IDE aesthetic cohorts, which include primary augmentation and revision. In the fourth quarter of 2021, the Company initiated a modular pre-market approval, or PMA, submission process with FDA and submitted the first of four expected modules. As of December 31, 2021, the Company is continuing to enroll subjects in the remaining reconstruction cohort.
2. Summary of Significant Accounting Policies
Basis of Presentation and Consolidation
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America, or GAAP, and the applicable rules and regulations of the Securities and Exchange Commission, or SEC.
F-8
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
The consolidated financial statements include the Company’s accounts and those of its wholly owned subsidiaries as of December 31, 2021 as follows:
| Subsidiary | Incorporation/Acquisition Date | ||||
| Establishment Labs, S.A. (Costa Rica) | January 18, 2004 | ||||
| Motiva USA, LLC (USA) | February 20, 2014 | ||||
| JAMM Technologies, Inc. (USA) | October 27, 2015 | ||||
| Establishment Labs Produtos par Saude Ltda (Brazil) | January 4, 2016 | ||||
| European Distribution Center Motiva BVBA (Belgium) | March 4, 2016 | ||||
| Motiva Implants France SAS (France) | September 12, 2016 | ||||
| JEN-Vault AG (Switzerland) | November 22, 2016 | ||||
| Motiva Nordica AB (Sweden) | November 2, 2017 | ||||
| Motiva Implants UK Limited (the United Kingdom) | July 31, 2018 | ||||
| Motiva Italy S.R.L (Italy) | July 31, 2018 | ||||
| Motiva Implants Spain, S.L. (Spain) | January 3, 2019 | ||||
| Motiva Austria GmbH (Austria) | January 14, 2019 | ||||
| Motiva Germany GmbH (Germany) | August 1, 2019 | ||||
| Motiva Argentina S.R.L. (Argentina) | February 7, 2020 | ||||
All intercompany accounts and transactions have been eliminated in consolidation.
Segments
The chief operating decision maker for the Company is the Chief Executive Officer. The Chief Executive Officer reviews financial information presented on a consolidated basis, accompanied by information about revenue by geographic region, for purposes of allocating resources and evaluating financial performance. The Company has one business activity and there are no segment managers who are held accountable for operations, operating results or plans for levels or components below the consolidated unit level. Accordingly, the Company has determined that it has a single reportable and operating segment structure. The Company and its Chief Executive Officer evaluate performance based primarily on revenue in the geographic regions in which the Company operates.
Geographic Concentrations
The Company derives all its revenues from sales to customers in Europe, the Middle East, Latin America, and Asia, and has not yet received approval to sell its products in the United States.
For the years ended December 31, 2021, 2020 and 2019, Brazil accounted for 11.6%, 10.9% and 15.7%, respectively, of consolidated revenue and no other individual country exceeded 10% of consolidated revenue, on a ship-to destination basis.
The majority of the Company’s consolidated total assets, including cash and tangible assets, is held in the United States. The Company’s long-lived assets, which primarily consist of property and equipment and intangible assets located in Costa Rica represented 84% and 80% of the total long-lived assets as of December 31, 2021 and 2020, respectively.
Use of Estimates
The preparation of financial statements in accordance with GAAP requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Significant accounting estimates and management judgments reflected in the consolidated financial statements include items such as accounts receivable valuation and allowances, inventory valuation and allowances,
F-9
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
valuation of acquired intangible assets, valuation of derivatives and valuation of deferred income tax assets, including tax valuation allowances. Estimates are based on historical experience, where applicable, and other assumptions believed to be reasonable by management. Actual results may differ from those estimates under different assumptions or conditions.
Concentration of Credit Risk and Other Risks and Uncertainties
Financial instruments that potentially subject the Company to a concentration of credit risk consist principally of cash and accounts receivable. The majority of the Company’s cash is held at two financial institutions in the United States. Balances in the Company’s cash accounts exceed the Federal Deposit Insurance Corporation, or FDIC, limit of $250,000. The Company has not experienced any losses to its deposits of cash.
All of the Company’s revenue has been derived from sales of its products in international markets, principally Europe, the Middle East, Latin America, and Asia. In the international markets in which the Company operates, the Company uses a combination of distributors and makes direct sales to customers. The Company performs ongoing credit evaluations of its distributors and customers, does not require collateral, and maintains allowances for potential credit losses on customer accounts when deemed necessary.
Substantially all of the Company’s revenues were derived from the sale of Motiva Implants. During the years ended December 31, 2021, 2020 and 2019, no customer accounted for more than 10% of the Company’s revenue. One customer accounted for 11.8% of the Company’s trade accounts receivable balance as of December 31, 2021. No customers accounted for more than 10% of the Company’s trade accounts receivable balance as of December 31, 2020.
The Company relies on NuSil Technology, LLC, or NuSil, as the sole supplier of medical-grade silicone used in Motiva Implants. During the years ended December 31, 2021, 2020 and 2019, the Company had purchases of $23.1 million, or 51.4% of total purchases, $15.3 million, or 66.7% of total purchases, and $14.2 million or 58.5% of total purchases, respectively, from NuSil. As of December 31, 2021 and 2020, the Company had an outstanding balance owed to this vendor of $2.5 million and $1.3 million, respectively.
The Company’s future results of operations involve a number of risks and uncertainties. Factors that could affect the Company’s future operating results and cause actual results to vary materially from expectations include, but are not limited to, uncertainty of regulatory approval of the Company’s current and potential future products, uncertainty of market acceptance of the Company’s products, competition from substitute products and larger companies, securing and protecting proprietary technology, access to capital, strategic relationships and dependence on key individuals and sole source suppliers.
Products developed by the Company require clearances from the FDA or other international regulatory agencies prior to commercial sales. There can be no assurance that the products will receive the necessary clearances. If the Company is denied clearance, clearance is delayed, or the Company is unable to maintain its existing clearances, these developments could have a material adverse impact on the Company.
The COVID-19 outbreak caused a material disruption of the operations of the Company and its suppliers and customers in fiscal 2020 and resulted in delayed clinical trial enrollment within the reconstruction cohort of its IDE clinical trial in the United States. However, to date, the impact from the COVID-19 outbreak has not had a material effect on the Company’s liquidity or financial position. The full extent of any future impact of the continuing outbreak, related business and travel restrictions, and changes to behavior intended to reduce its spread is uncertain and continues to evolve globally. Management continues to monitor the impact that the COVID-19 pandemic is having on the Company, the breast aesthetics and reconstruction market and the economies in which the Company operates.
Cash
The Company’s cash consists of cash maintained in checking and interest-bearing accounts. The majority of the Company’s cash is held at two financial institutions in the United States. The Company accounts for financial instruments with original maturities of three months or less at the date of purchase as cash equivalents. The Company held no cash equivalents as of December 31, 2021 and 2020.
F-10
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
Accounts Receivable and Allowance for Doubtful Accounts
Accounts receivable is stated at invoice value less estimated allowances for returns and doubtful accounts. The Company continually monitors customer payments and maintains an allowance for estimated losses resulting from customers’ inability to make required payments. In evaluating the Company’s ability to collect outstanding receivable balances, the Company considers various factors including the age of the balance, the creditworthiness of the customer, which is assessed based on ongoing credit evaluations and payment history, and the customer’s current financial condition. In cases where there are circumstances that may impair a specific customer’s ability to meet its financial obligations, an allowance is recorded against amounts due, which reduces the net recognized receivable to the amount reasonably believed to be collectible.
A roll-forward of the allowance for doubtful accounts is as follows:
| Year Ended December 31, | ||||||||||||||||||||
| 2021 | 2020 | 2019 | ||||||||||||||||||
| Beginning balance | $ | 1,143 | $ | 1,026 | $ | 926 | ||||||||||||||
| Provision for doubtful accounts | 200 | 375 | 111 | |||||||||||||||||
| Write-offs | (122) | (258) | (11) | |||||||||||||||||
| Ending balance | $ | 1,221 | $ | 1,143 | $ | 1,026 | ||||||||||||||
Inventory and Cost of Revenue
Inventory is stated at the lower of cost to purchase or manufacture the inventory or the net realizable value of such inventory. Cost is determined using the standard cost method which approximates actual costs using the first-in, first-out basis. The Company regularly reviews inventory quantities considering actual losses, projected future demand, and remaining shelf life to record a provision for excess and slow-moving inventory.
A roll-forward of the inventory reserve is as follows:
| Year Ended December 31, | ||||||||||||||||||||
| 2021 | 2020 | 2019 | ||||||||||||||||||
| Beginning balance | $ | 1,625 | $ | 347 | $ | 230 | ||||||||||||||
| Provision for inventory obsolescence | 338 | 1,180 | 143 | |||||||||||||||||
| Write-offs | (796) | 98 | (26) | |||||||||||||||||
| Ending balance | $ | 1,167 | $ | 1,625 | $ | 347 | ||||||||||||||
The Company recognizes the cost of inventory transferred to the customer in cost of revenue when revenue is recognized.
Leases
The Company determines if an arrangement is, or contains, a lease at the inception date of the contract. The Company has elected an expedient to account for each separate lease component and its associated non-lease components as a single lease component for the majority of its asset classes.
The lease term may include periods covered by options to extend or terminate the lease when it is reasonably certain that the Company will exercise a renewal option, or reasonably certain it will not exercise an early termination option. The Company recognizes lease liabilities and right-of-use, or ROU, assets upon commencement for all material leases with a term greater than 12 months. The Company has elected an
F-11
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
expedient not to recognize leases with a lease term of 12 months or less on the balance sheet. These short-term leases are expensed on a straight-line basis over the lease term.
Shipping and Handling Costs
Shipping and handling costs are expensed as incurred and are included in selling, general and administrative, or SG&A, expenses. For the years ended December 31, 2021, 2020 and 2019, shipping and handling costs were $5.3 million, $3.2 million and $3.2 million, respectively.
Revenue Recognition
The Company recognizes revenue related to sales of products to distributors or directly to customers in markets where it has regulatory approval, net of discounts and allowances. The Company recognizes revenue in accordance with Accounting Standards Codification, or ASC, Revenue from Contracts with Customers (Topic 606). ASC 606 requires the Company to recognize revenue to depict the transfer of goods or services to a customer at an amount that reflects the consideration it expects to receive in exchange for those goods or services.
The Company recognizes revenue related to the sales of products to distributors at the time of shipment of the product, which represents the point in time when the distributor has taken ownership and assumed the risk of loss, and the required revenue recognition criteria are satisfied. The Company’s distributors are obligated to pay within specified terms regardless of when, or if, they sell the products. The Company’s contracts with distributors typically do not contain right of return or price protection and have no post-delivery obligations.
The Company recognizes revenue when title to the product and risk of loss transfer to customers, provided there are no remaining performance obligations required of the Company or any written matters requiring customer acceptance. The Company allows for the return of product from direct customers in certain regions in limited instances within fifteen days after the original sale and records estimated sales returns as a reduction of sales in the same period revenue is recognized. Appropriate reserves are established for anticipated sales returns based on historical experience, recent gross sales and any notification of pending returns. Actual sales returns in any future period are inherently uncertain and thus may differ from the estimates. If actual sales returns differ significantly from the estimates, an adjustment to revenue in the current or subsequent period is recorded. As of December 31, 2021, 2020 and 2019, an allowance of $10,000, $54,000 and $36,000 was recorded for product returns, respectively.
A portion of the Company’s revenue is generated from the sale of consigned inventory maintained at physician, hospital, or clinic locations. For these products, revenue is recognized at the time the Company is notified by the consignee that the product has been implanted, not when the consigned products are delivered to the consignee’s warehouse.
Revenue was generated in these primary geographic markets:
| Year Ended December 31, | ||||||||||||||||||||
| 2021 | 2020 | 2019 | ||||||||||||||||||
(in thousands) | ||||||||||||||||||||
| Europe | $ | 51,912 | $ | 37,667 | $ | 36,212 | ||||||||||||||
| Latin America | 38,226 | 21,512 | 27,994 | |||||||||||||||||
| Asia-Pacific/Middle East | 35,679 | 24,986 | 24,819 | |||||||||||||||||
| Other | 865 | 511 | 540 | |||||||||||||||||
| $ | 126,682 | $ | 84,676 | $ | 89,565 | |||||||||||||||
The Company has a limited warranty for the shelf life of breast implants, which is five years from the time of manufacture. Estimated warranty obligations are recorded at the time of sale. The Company also offers a
F-12
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
warranty to patients in the event of rupture and a replacement program for capsular contracture events, provided certain registration requirements are met. Revenue for extended warranties is recognized ratably over the term of the agreement. To date, these warranty and program costs have been de minimis. The Company will continue to evaluate the warranty reserve policies for adequacy considering claims history.
Deferred revenue primarily consists of payments received in advance of meeting revenue recognition criteria. The Company has received payments from distributors to provide distribution exclusivity within a geographic area and recognizes deferred revenue on a ratable basis over the term of such contractual distribution relationship. Additionally, the Company has received payments from customers in direct markets prior to surgical implantation and recognizes deferred revenue at the time the Company is notified by the customer that the product has been implanted. For all arrangements, any revenue that has been deferred and is expected to be recognized beyond one year is classified as long-term deferred revenue and included in “Other liabilities, long-term” on the consolidated balance sheets (see Note 3).
Research and Development
Costs related to research and development, or R&D, activities are expensed as incurred. R&D costs primarily include personnel costs, materials, clinical expenses, regulatory expenses, product development, consulting services, and outside research activities, all of which are directly related to research and development activities.
The Company estimates IDE clinical trial expenses based on the services performed, pursuant to contracts with research institutions and clinical research organizations that conduct and manage clinical trials on its behalf. In accruing service fees, the Company estimates the time period over which services will be performed and the level of patient enrollment and activity expended in each period. If the actual timing of the performance of services or the level of effort varies from the estimate, the Company will adjust the accrual accordingly.
Selling, General and Administrative Expenses
SG&A expenses include sales and marketing costs, payroll and related benefit costs, insurance expenses, shipping and handling costs, legal and professional fees and administrative overhead.
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation and amortization.
Following the exercise of its option to purchase its manufacturing facility in June 2019, the Company depreciates the owned building on a straight-line basis over 50 years of useful life. Depreciation of property and equipment is computed using the straight-line method over the assets’ estimated useful lives of to ten years. Leasehold improvements are amortized on a straight-line basis over the shorter of the estimated useful life of the asset or the remaining lease term after factoring expected renewal periods. Upon retirement or disposal of assets, the costs and related accumulated depreciation are eliminated from the accounts and any gain or loss is recognized in operations. Maintenance and repairs are expensed as incurred. Substantially all of the Company’s manufacturing operations and related property and equipment is located in Costa Rica.
Goodwill and Intangible Assets
The Company records the excess of the acquisition purchase price over the net fair value of the tangible and identifiable intangible assets acquired and liabilities assumed as goodwill. In accordance with ASC 350, Intangibles - Goodwill and Other, the Company tests goodwill for impairment annually during the fourth quarter of each year and whenever events or changes in circumstances indicate that the carrying value of the asset may not be recoverable. In connection with the annual impairment test for goodwill, the Company elected the option to perform a qualitative assessment to determine whether it is more likely than not that the fair value of the reporting unit is less than its carrying amount. If the Company determines that it was more likely than not that the fair value of the reporting unit is less than its carrying amount, then the impairment test is performed.
Consistent with the Company's assessment that it has only one reporting segment, the Company has determined that it has only one reporting unit and tests goodwill for impairment at the entity level using the two-step process required by ASC 350. In the first step, the Company compares the carrying amount of the reporting unit to the fair value of the enterprise. If the fair value of the enterprise exceeds the carrying value, goodwill is not considered impaired and no further testing is required. If the carrying value of the enterprise exceeds the fair value, goodwill
F-13
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
is potentially impaired, and the second step of the impairment test must be performed. In the second step, the Company compares the implied fair value of the goodwill, as defined by ASC 350, to its carrying amount to determine the impairment loss, if any.
The Company capitalizes certain costs related to intangible assets, such as patents, trademarks and software development costs. The Company follows the provisions of ASC 350-40, Internal Use Software for determining whether computer software is internal-use software and on accounting for the proceeds of computer software originally developed or obtained for internal use. The Company expenses all costs incurred during the preliminary project stage of software development and capitalizes the costs incurred during the application development stage. Costs incurred relating to upgrades and enhancements to the software are capitalized if it is determined that these upgrades or enhancements add additional functionality to the software. Costs incurred to improve and support products after they become available are charged to expense as incurred.
The Company records purchased intangible assets at their respective estimated fair values at the date of acquisition. Purchased finite-lived intangible assets are being amortized using the straight-line method over their remaining estimated useful lives, which range from to fifteen years. The Company evaluates the remaining useful lives of intangible assets on a periodic basis to determine whether events or circumstances warrant a revision to the remaining estimated amortization period. The Company tests indefinite-lived intangible assets for impairment on at least an annual basis and whenever circumstances suggest the assets may be impaired. If indicators of impairment are present, the Company evaluates the carrying value of the intangible assets in relation to estimates of future undiscounted cash flows. The Company also evaluates the remaining useful life of an indefinite-lived intangible asset to determine whether events and circumstances continue to support an indefinite useful life.
During the years ended December 31, 2021, 2020 and 2019, there has been no impairment of goodwill or intangible assets based on the qualitative assessments performed by the Company.
Long-Lived Assets
The Company reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset might not be recoverable. When such an event occurs, management determines whether there has been impairment by comparing the anticipated undiscounted future net cash flows to the related asset group’s carrying value. If an asset is considered impaired, the asset is written down to fair value, which is determined based either on discounted cash flows or appraised value, depending on the nature of the asset. There were no impairment charges, or changes in estimated useful lives recorded during the years ended December 31, 2021, 2020 and 2019.
Debt and Embedded Derivatives
The Company applies the accounting standards for derivatives and for distinguishing liabilities from equity when accounting for hybrid contracts. The Company accounts for convertible debt instruments when the Company has determined that the embedded conversion options should not be bifurcated from their host instruments in accordance with ASC 470-20 Debt with Conversion and Other Options (see Note 6).
The Company uses option pricing valuation models to determine the fair value of embedded derivatives and records any change in fair value as a component of other income or expense in the consolidated statements of operations (see Note 5).
Debt Issuance Costs and Debt Discounts
Costs incurred in connection with the issuance of new debt are capitalized. Capitalizable debt issuance costs paid to third parties and debt discounts, net of amortization, are recorded as a reduction to the long-term debt balance on the consolidated balance sheets. Amortization expense on capitalized debt issuance costs and debt discounts related to loans are calculated using the effective interest method over the term of the loan commitment and is recorded as interest expense in the consolidated statements of operations.
Income Taxes
The Company records income taxes using the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized
F-14
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
in the Company’s consolidated financial statements or income tax returns. In estimating future tax consequences, expected future events, enactments or changes in the tax law or rates are considered. Valuation allowances are provided when necessary to reduce deferred tax assets to the amount expected to be realized.
The Company operates in various tax jurisdictions and is subject to audit by various tax authorities.
The Company records uncertain tax positions based on a two-step process whereby (1) a determination is made as to whether it is more likely than not that the tax positions will be sustained based on the technical merits of the position and (2) for those tax positions that meet the more-likely-than-not recognition threshold the Company recognizes the largest amount of tax benefit that is greater than 50% likely to be realized upon ultimate settlement with the related tax authority. The Company’s policy is to recognize interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense. Significant judgment is required in the identification of uncertain tax positions and in the estimation of penalties and interest on uncertain tax positions.
There were no material uncertain tax positions in the years ended December 31, 2021, 2020 and 2019.
Foreign Currency
The financial statements of the Company’s foreign subsidiaries whose functional currencies are the local currencies are translated into U.S. dollars for consolidation as follows: assets and liabilities at the exchange rate as of the balance sheet date, stockholders’ equity at the historical rates of exchange, and income and expense amounts at the average exchange rate for the period. Translation adjustments resulting from the translation of the subsidiaries’ accounts are included in “Accumulated other comprehensive income” as equity in the consolidated balance sheet. Transactions denominated in currencies other than the applicable functional currency are converted to the functional currency at the exchange rate on the transaction date. At period end, monetary assets and liabilities are remeasured to the functional currency using exchange rates in effect at the balance sheet date. Non-monetary assets and liabilities are remeasured at historical exchange rates. Gains and losses resulting from foreign currency transactions are included within “Other income (expense), net” in the consolidated statement of operations. For the years ended December 31, 2021, 2020 and 2019, foreign currency transaction loss amounted to $5.6 million, $1.7 million and $1.2 million, respectively.
Comprehensive Loss
The Company’s comprehensive loss consists of net loss and foreign currency translation adjustments arising from the consolidation of the Company’s foreign subsidiaries.
Share-Based Compensation
The Company measures and recognizes compensation expense for all share-based awards in accordance with the provisions of ASC 718, Stock Compensation. Share-based awards granted include stock options, restricted stock units, or RSUs, and restricted stock awards, or RSAs. Share-based compensation expense for stock options and restricted stock granted to employees is measured at the grant date based on the fair value of the awards and is recognized as an expense ratably on a straight-line basis over the requisite service period. The fair value of options to purchase shares granted to employees is estimated on the grant date using the Black-Scholes option valuation model.
The calculation of share-based compensation expense requires the Company to make assumptions and judgments about the variables used in the Black-Scholes model, including the expected term, expected volatility of the underlying common shares, risk-free interest rate and dividends.
Net Income (Loss) Per Share
Basic net income (loss) per share is calculated by dividing the net income (loss) attributable to shareholders by the weighted-average number of shares outstanding during the period, without consideration for potentially dilutive securities. Diluted net income (loss) per share is computed by dividing the net income (loss) by the weighted-average number of shares and potentially dilutive securities outstanding for the period. For purposes of the diluted net loss per share calculation, any shares issuable upon exercise of warrants, stock options and non-vested restricted stock outstanding under the Company’s equity plan are potentially dilutive securities. Diluted net loss per share is the same as basic net loss per share for periods where the Company reported a net loss because including the dilutive securities would be anti-dilutive.
F-15
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
Reclassifications
Certain reclassifications have been made to prior year amounts to conform to the current year presentation. These reclassifications had no material impact on the Company’s financial position or results of operations for the year ended December 31, 2021.
Recent Accounting Standards
Periodically, new accounting pronouncements are issued by the Financial Accounting Standards Board, or FASB, or other standard setting bodies and adopted by the Company as of the specified effective date. Unless otherwise discussed, the impact of recently issued standards that are not yet effective will not have a material impact on the Company’s consolidated financial statements upon adoption. Previously, under the Jumpstart Our Business Startups Act of 2012, or JOBS Act, the Company met the definition of an emerging growth company, and previously elected the extended transition period for complying with new or revised accounting standards pursuant to Section 107(b) of the JOBS Act. The Company ceased to be an emerging growth company on December 31, 2021.
The following recent accounting pronouncements issued by the FASB, could have a material effect on the Company’s financial statements:
Recently Adopted Accounting Standards
In December 2019, the FASB issued ASU No. 2019-12, Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes, as part of its initiative to reduce complexity in the accounting standards. The standard eliminates certain exceptions related to the approach for intra-period tax allocation, the methodology for calculating income taxes in an interim period and the recognition of deferred tax liabilities for outside basis differences. The standard also clarifies and simplifies other aspects of the accounting for income taxes. This standard is currently effective. The Company adopted ASU 2019-12 as of September 30, 2021. The adoption did not have a material impact upon the Company’s financial position and results of operations.
In August 2018, the FASB issued ASU No. 2018-13, Fair Value Measurement: Disclosure Framework - Changes to the Disclosure Requirements for Fair Value Measurement. This ASU modifies the disclosure requirements for fair value measurements. The modifications removed the following disclosure requirements: (i) the amount of, and reasons for, transfers between Level 1 and Level 2 of the fair value hierarchy; (ii) the policy for timing of transfers between levels; and (iii) the valuation processes for Level 3 fair value measurements. This ASU added the following disclosure requirements: (i) the changes in unrealized gains and losses for the period included in other comprehensive income, or OCI, for recurring Level 3 fair value measurements held at the end of the reporting period; and (ii) the range and weighted average of significant observable inputs used to develop Level 3 fair value measurements. This standard is currently effective. The Company adopted ASU 2018-13 on January 1, 2021. As the requirements of this literature are disclosure only, ASU 2018-13 did not impact the Company’s financial condition or results of operations.
In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments - Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments. This ASU replaces the impairment methodology in current GAAP, which delays recognition of credit losses until it is probable a loss has been incurred, with a methodology that reflects expected credit losses and requires consideration of a broader range of reasonable and supportable information to inform credit loss estimates. This standard is currently effective. During the fourth quarter of 2021, the Company adopted ASU 2016-13 as of January 1, 2021 using the modified retrospective method. The adoption did not have a material impact upon the Company’s financial position and results of operations.
Recently Issued Accounting Standards
In August 2020, the FASB issued ASU No. 2020-06, Debt with Conversion and other Options (Subtopic 470-20) and Derivatives and Hedging - Contracts in Entity’s Own Equity (Subtopic 815-40). The new guidance eliminates the beneficial conversion and cash conversion accounting models for convertible instruments. It also amends the accounting for certain contracts in an entity’s own equity that are currently accounted for as derivatives because of specific settlement provisions. In addition, the new guidance modifies how particular convertible instruments and certain contracts that may be settled in cash or shares impact the diluted EPS computation. The standard is effective for fiscal years beginning after December 15, 2021, including interim periods within those fiscal years.
F-16
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
The Company is currently evaluating the effect the updated standard will have on its consolidated financial statements and footnote disclosures.
3. Balance Sheet Accounts
Inventory, Net
| December 31, | ||||||||||||||
| 2021 | 2020 | |||||||||||||
(in thousands) | ||||||||||||||
| Raw materials | $ | 8,519 | $ | 5,450 | ||||||||||
| Work in process | 1,396 | 1,121 | ||||||||||||
| Finished goods | 18,492 | 16,639 | ||||||||||||
| $ | 28,407 | $ | 23,210 | |||||||||||
As of December 31, 2021 and 2020, $3.5 million and $2.0 million of inventory was on consignment, respectively.
Prepaid Expenses and Other Current Assets
| December 31, | ||||||||||||||
| 2021 | 2020 | |||||||||||||
(in thousands) | ||||||||||||||
| Prepaid insurance | $ | 2,315 | $ | 2,115 | ||||||||||
| Prepaid raw materials and accessories | 577 | 164 | ||||||||||||
| Prepaid warranty and distribution rights | 516 | 486 | ||||||||||||
| Prepaid US clinical trial costs | 412 | 57 | ||||||||||||
| Prepaid taxes | 551 | 528 | ||||||||||||
| 2,641 | 2,089 | |||||||||||||
| $ | 7,012 | $ | 5,439 | |||||||||||
F-17
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
Property and Equipment, Net
| December 31, | ||||||||||||||
| 2021 | 2020 | |||||||||||||
(in thousands) | ||||||||||||||
| Machinery and equipment | $ | 10,240 | $ | 9,232 | ||||||||||
| Building improvements | 6,713 | 6,456 | ||||||||||||
| Furniture and fixtures | 4,761 | 4,092 | ||||||||||||
| Building | 2,472 | 2,472 | ||||||||||||
| Leasehold improvements | 2,118 | 2,065 | ||||||||||||
| Land | 802 | 802 | ||||||||||||
| Vehicles | 268 | 399 | ||||||||||||
| Construction in process | 3,174 | 317 | ||||||||||||
| Total | 30,548 | 25,835 | ||||||||||||
| Less: Accumulated depreciation and amortization | (11,890) | (9,633) | ||||||||||||
| $ | 18,658 | $ | 16,202 | |||||||||||
For the years ended December 31, 2021, 2020 and 2019, depreciation and amortization expense related to property and equipment was $2.5 million, $2.4 million and $2.7 million, respectively.
The Company entered into finance leases relating to equipment and vehicles and recorded the fair value of the lease payments on the initial contract date and is amortizing the assets over the term of the leases. As of each of December 31, 2021 and 2020, the gross asset value for finance lease assets was $1.4 million. Depreciation expense for assets under finance leases was $126,000, $80,000 and $84,000 for the years ended December 31, 2021, 2020 and 2019, respectively.
In August 2021, the Company entered into a contract with the Zona Franca Coyol, S.A., or CFZ, to begin construction of a new manufacturing facility in Costa Rica. The costs for improvement of the land and construction of a cold shell building are being paid for by CFZ and, upon completion, the Company will have the option to purchase the title to the land and cold shell building for approximately $12.6 million or to lease the facility at a to be determined price. Subject to purchase of the land and cold shell building, the Company will have the option to buy an adjacent lot of land for approximately $2.8 million and engage CFZ to construct an additional manufacturing facility.
F-18
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
Accrued Liabilities
Accrued liabilities consisted of the following:
| December 31, | ||||||||||||||
| 2021 | 2020 | |||||||||||||
(in thousands) | ||||||||||||||
| Performance bonus | $ | 3,346 | $ | 2,406 | ||||||||||
| Payroll and related expenses | 3,904 | 2,781 | ||||||||||||
| Bonus feature of stock option grants | 5,570 | 5,992 | ||||||||||||
| 402 | 788 | |||||||||||||
| Commissions | 1,138 | 628 | ||||||||||||
| Professional and legal services | 819 | 439 | ||||||||||||
| Warranty reserve | 167 | 237 | ||||||||||||
| Cash payable for asset acquisitions - contingent consideration | 137 | 147 | ||||||||||||
| 753 | 1,114 | |||||||||||||
| $ | 16,236 | $ | 14,532 | |||||||||||
Other Liabilities, Short-Term
Other liabilities, short-term consisted of the following:
| December 31, | ||||||||||||||
| 2021 | 2020 | |||||||||||||
(in thousands) | ||||||||||||||
| Deferred revenue | 769 | 1,214 | ||||||||||||
| Cash payable for asset acquisitions | 409 | 432 | ||||||||||||
| $ | 1,178 | $ | 1,646 | |||||||||||
Other Liabilities, Long-Term
Other liabilities, long-term consisted of the following:
| December 31, | ||||||||||||||
| 2021 | 2020 | |||||||||||||
(in thousands) | ||||||||||||||
| Deferred revenue | $ | 2,392 | $ | 1,860 | ||||||||||
| Cash payable for asset acquisitions | — | 425 | ||||||||||||
| — | 47 | |||||||||||||
| $ | 2,392 | $ | 2,332 | |||||||||||
F-19
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
4. Goodwill and Other Intangible Assets
Goodwill represents the excess of the purchase price over the fair value of the net tangible and identifiable intangible assets acquired in a business combination. Intangible assets resulting from the acquisitions of entities accounted for using the acquisition method of accounting are recorded at the estimated fair value of the assets acquired. Purchased intangibles include certain patents and license rights, 510(k) authorization by the FDA to sell a medical device and other intangible assets.
The Company’s goodwill and most intangibles at December 31, 2021 are the result of previous asset and business acquisitions. Finite-lived intangibles are amortized over their estimated useful lives based on expected future benefit.
In addition to the intangibles acquired, the Company capitalized certain patent and license rights as identified intangibles based on patent and license rights agreements entered into over the past several years. Additionally, the Company capitalized certain software development costs.
There were no changes in the carrying amount of goodwill during the year ended December 31, 2021:
| Balance as of January 1, 2021 | Additions | Accumulated Impairment Losses | Balance as of December 31, 2021 | ||||||||||||||||||||
| (in thousands) | |||||||||||||||||||||||
| Goodwill | $ | 465 | $ | — | $ | — | $ | 465 | |||||||||||||||
The carrying amounts of these intangible assets other than goodwill as of December 31, 2021 were as follows:
| Gross Carrying Amount | Accumulated Amortization | Net Carrying Amount | Estimated Useful Lives | ||||||||||||||||||||
| (in thousands) | (in years) | ||||||||||||||||||||||
| Patents and license rights | $ | 1,736 | $ | (1,136) | $ | 600 | 7-12 | ||||||||||||||||
| Customer relationships | 2,033 | (1,799) | 234 | 4-10 | |||||||||||||||||||
| 510(k) authorization | 567 | (232) | 335 | 15 | |||||||||||||||||||
| Developed technology | 62 | (52) | 10 | 10 | |||||||||||||||||||
| Capitalized software development costs | 3,648 | (791) | 2,857 | 2-5 | |||||||||||||||||||
| Other | 75 | (31) | 44 | 2-5 | |||||||||||||||||||
| Capitalized patents and license rights not yet amortized | 291 | — | 291 | ||||||||||||||||||||
| $ | 8,412 | $ | (4,041) | $ | 4,371 | ||||||||||||||||||
F-20
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
The carrying amounts of intangible assets other than goodwill as of December 31, 2020 were as follows:
| Gross Carrying Amount | Accumulated Amortization | Net Carrying Amount | Estimated Useful Lives | ||||||||||||||||||||
| (in thousands) | (in years) | ||||||||||||||||||||||
| Patents and license rights | $ | 1,736 | $ | (951) | $ | 785 | 7-12 | ||||||||||||||||
| Customer relationships | 2,033 | (1,297) | 736 | 4-10 | |||||||||||||||||||
| 510(k) authorization | 567 | (194) | 373 | 15 | |||||||||||||||||||
| Developed technology | 62 | (46) | 16 | 10 | |||||||||||||||||||
| Capitalized software development costs | 2,203 | (302) | 1,901 | 2-5 | |||||||||||||||||||
| Other | 75 | (29) | 46 | 2-5 | |||||||||||||||||||
| Capitalized patents and license rights not yet amortized | 291 | — | 291 | ||||||||||||||||||||
| $ | 6,967 | $ | (2,819) | $ | 4,148 | ||||||||||||||||||
The amortization expense associated with intangible assets was $1.2 million, $0.9 million and $0.6 million for the years ended December 31, 2021, 2020 and 2019, respectively. Non-product related amortization is recorded in SG&A while product related amortization is recorded in cost of revenue.
As of December 31, 2021, the amortization expense related to identifiable intangible assets, with definite useful lives, in future periods is expected to be as follows:
| Year Ending December 31, | (in thousands) | |||||||
| 2022 | $ | 1,111 | ||||||
| 2023 | 859 | |||||||
| 2024 | 784 | |||||||
| 2025 | 617 | |||||||
| 2026 | 330 | |||||||
| Thereafter | 379 | |||||||
| Total | $ | 4,080 | ||||||
The Company evaluates the recoverability of goodwill and indefinite-lived intangible assets annually and whenever events or changes in circumstances indicate that the carrying amount may not be recoverable. As of December 31, 2021, there has been no impairment of goodwill or intangible assets based on the qualitative assessments performed by the Company.
5. Fair Value Measurements
The carrying value of the Company’s cash, accounts receivable and accounts payable approximate fair value due to the short-term nature of these items. Embedded derivatives that qualify for liability treatment are carried at fair value and re-measured at each reporting period.
Fair value is defined as the exchange price that would be received for an asset or an exit price paid to transfer a liability in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs.
F-21
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
The fair value hierarchy defines a three-level valuation hierarchy for disclosure of fair value measurements as follows:
▪Level I Unadjusted quoted prices in active markets for identical assets or liabilities;
▪Level II Inputs other than quoted prices included within Level I that are observable, unadjusted quoted prices in markets that are not active, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the related assets or liabilities; and
▪Level III Unobservable inputs that are supported by little or no market activity for the related assets or liabilities.
The categorization of a financial instrument within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement.
The following table sets forth the Company’s financial instruments that were measured at fair value on a recurring basis by level within the fair value hierarchy at period end:
| Fair Value Measurements at December 31, 2021 | |||||||||||||||||||||||
| Total | Level 1 | Level 2 | Level 3 | ||||||||||||||||||||
| (in thousands) | |||||||||||||||||||||||
| Liabilities | |||||||||||||||||||||||
| Madryn put option liability | $ | 703 | $ | — | $ | — | $ | 703 | |||||||||||||||
| $ | 703 | $ | — | $ | — | $ | 703 | ||||||||||||||||
| Fair Value Measurements at December 31, 2020 | |||||||||||||||||||||||
| Total | Level 1 | Level 2 | Level 3 | ||||||||||||||||||||
| (in thousands) | |||||||||||||||||||||||
| Liabilities | |||||||||||||||||||||||
| Madryn put option liability | $ | 1,440 | $ | — | $ | — | $ | 1,440 | |||||||||||||||
| $ | 1,440 | $ | — | $ | — | $ | 1,440 | ||||||||||||||||
The fair value measurement of derivatives is based on significant inputs not observed in the market and thus represents a Level 3 measurement.
In August 2017, the Company entered into a credit agreement, or the Madryn Credit Agreement, with Madryn Health Partners, LP, or Madryn, as administrative agent, and a syndicate of lenders (see Note 6). The Company determined that the Madryn Credit Agreement contained put options related to early redemption mandatory prepayment terms in case of change in control or an event of default and a call option related to voluntary repayment option. The Company allocated a fair value of $15.1 million for these identified embedded derivatives as a debt discount on the original commitment date. An additional $5.0 million and $1.6 million debt discount was recorded on respective borrowing dates when the Company met the required milestones and borrowed an additional $10.0 million in 2017 and $25.0 million in 2019. The Company revalued the options as of each reporting period and recorded the change in the fair value in the consolidated statement of operations as other income or expense.
Valuation of the embedded derivatives is complex and requires interest rate simulation, capturing optimal decision making process as interest rate fluctuates, and estimating the resultant bond valuation and the resultant pay-off to the option holder. The Company estimated the fair value of the embedded redemption options based on a “with” and “without” approach using the Black-Derman-Toy model, a form of the Binomial Lattice Model that captures
F-22
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
interest rate variability and the prepayment optionality. The Binomial Lattice Model allows for the possibility of exercise before the end of the option’s life and considers future interest rates, volatility and other data with regards to the Company’s credit rating and credit spread. The value of the embedded derivatives was based on the difference between the “with” and “without” analysis. The probability of a change in control occurring was determined to be 50% (cumulative probability through the maturity date) at December 31, 2021 and December 31, 2020.
The Company used the following assumptions to value Madryn derivatives:
Madryn Put Option Liability
| December 31, | |||||||||||
| 2021 | 2020 | ||||||||||
| Interest rate volatility | 25.8% | 19.7% | |||||||||
| Market yield rate | 6.8% | 7.9% | |||||||||
| Term (in years) | 3.75 | 4.82 | |||||||||
| Dividend yield | —% | —% | |||||||||
The estimates are based, in part, on subjective assumptions and could differ materially in the future.
During the periods presented, the Company has not changed the manner in which it values liabilities that are measured at fair value using Level 3 inputs. The Company recognizes transfers between levels of the fair value hierarchy as of the end of the reporting period. There were no transfers within the hierarchy during the years ended December 31, 2021 and 2020.
The fair value of the debt redemption feature liability includes the estimated market rate (credit spread and risk-free rate) and volatility. The higher/lower the estimated volatility, the higher/lower the value of the debt redemption feature liability. The higher/lower the estimated market rate, the higher/lower the value of the debt redemption feature liability.
The following table sets forth a summary of the changes in the fair value of the Company’s Level 3 financial instruments as follows:
| Acquisition-related Contingent Consideration | Put Option Liability | |||||||||||||
| Balance at December 31, 2019 | $ | 922 | $ | 3,072 | ||||||||||
| Change in fair value | (304) | (1,632) | ||||||||||||
| Settlement | (618) | — | ||||||||||||
| Balance at December 31, 2020 | — | 1,440 | ||||||||||||
| Change in fair value | — | (737) | ||||||||||||
| Balance at December 31, 2021 | $ | — | $ | 703 | ||||||||||
6. Debt
Madryn Debt
On August 24, 2017, the Company entered into the Madryn Credit Agreement with Madryn, as administrative agent, and a syndicate of lenders that matures September 30, 2025. On August 5, 2020, the Company amended the Madryn Credit Agreement to adjust the minimum product revenue milestone previously applicable to December 31, 2020 to September 30, 2021 and to add Motiva Implants UK Limited, Motiva Implants France SAS,
F-23
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
Motiva Implants Spain, S.L. and Motiva Germany GmbH, wholly-owned subsidiaries of the Company, as guarantors to the Madryn Credit Agreement.
The Madryn Credit Agreement, as amended, provides for term loans in a maximum aggregate principal amount of $65.0 million.
In connection with the Madryn Credit Agreement, the Company and certain of its subsidiaries granted a security interest in substantially all of their respective assets, including, without limitation, intellectual property, and pledges of certain shares of the Company’s subsidiaries, subject to certain excluded collateral exceptions.
The Madryn Credit Agreement contains customary affirmative and negative covenants, including, but not limited to, restrictions on the ability of the Company and its subsidiaries to incur additional indebtedness, create liens, make certain investments, make restricted payments, enter into or undertake certain liquidations, mergers, consolidations or acquisitions and dispose of assets or subsidiaries. In addition, the Madryn Credit Agreement requires the Company to maintain minimum revenues and liquidity.
Borrowings under the Madryn Credit Agreement bear interest at a rate equal to 3-month LIBOR plus 8.0% per annum provided that no default has occurred. In an event of a default, the interest would increase by an additional 4.0% per annum. The effective interest rate under the amended Madryn Credit Agreement is 18.4%, and the weighted average interest rate was approximately 10.6% at December 31, 2021. The Company incurred $6.9 million, $7.6 million and $6.2 million in interest expense in connection with Madryn Credit Agreement during the years ended December 31, 2021, 2020 and 2019, respectively, including $0.7 million and $0.3 million of direct costs to amend the Madryn Credit Agreement in August 2020 and June 2019 which was expensed as interest expense. No principal payments are due on the term loans until the final maturity date on September 30, 2025.
The Company also determined that the Madryn Credit Agreement contained put options which are mandatory repayment provisions related to liquidity events or an event of default and a call option related to voluntary repayment option. The Company allocated a fair value of $15.1 million for these embedded derivatives as a debt discount on the original commitment date in August 2017. An additional $5.0 million and $1.6 million debt discount was recorded on respective borrowing dates when the Company met the required milestones and borrowed an additional $10.0 million in the fourth quarter of fiscal 2017 and $25.0 million in August 2019. The Company revalues the embedded derivatives as of each reporting period and records the change in the fair value in the consolidated statement of operations as other income or expense (see Note 5).
The Company recorded Madryn debt on the balance sheet as follows:
| December 31, | |||||||||||
| 2021 | 2020 | ||||||||||
| (in thousands) | |||||||||||
| Principal | $ | 65,000 | $ | 65,000 | |||||||
| Net unamortized debt discount and issuance costs | (13,094) | (15,168) | |||||||||
| Net carrying value of Madryn debt | $ | 51,906 | $ | 49,832 | |||||||
As of December 31, 2021, the Company is in compliance with all financial debt covenants.
7. Leases
The Company recognizes lease liabilities and ROU assets upon commencement for all material leases with a term greater than 12 months. The Company has elected an expedient not to recognize leases with a lease term of 12 months or less on the balance sheet. These short-term leases are expensed on a straight-line basis over the lease term.
F-24
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
ROU assets represent the Company’s right to use an underlying asset for the lease term and lease liabilities represent the Company’s obligation to make lease payments arising from the lease. ROU assets and lease liabilities are recognized at commencement date of the lease based on the present value of lease payments over the lease term. When the rate implicit to the lease cannot be readily determined, the Company utilizes its incremental borrowing rate in determining the present value of the future lease payments. Lease liabilities are accreted each period and reduced for payments. The ROU asset also includes other adjustments, such as for the effects of escalating rents, rent abatements or initial lease costs. The lease term may include periods covered by options to extend or terminate the lease when it is reasonably certain that the Company will exercise a renewal option, or reasonably certain it will not exercise an early termination option. For operating leases, lease expense for minimum lease payments is recognized on a straight-line basis over the expected lease term. For finance leases, the ROU asset depreciates on a straight-line basis over the shorter of the lease term or useful life of the ROU asset and the lease liability accretes interest based on the interest method using the discount rate determined at lease commencement.
The Company has operating leases for facilities and office space as well as finance leases for equipment and vehicles. Operating lease assets and the related lease liabilities are included within the ROU assets—operating leases. The determination of whether an arrangement is, or contains, a lease is performed at the inception of the arrangement. The Company has operating and finance leases for certain facilities, office space, equipment, and vehicles to be used in its operations, with remaining lease terms ranging from monthly to 7 years. These leases require monthly lease payments that may be subject to annual increases throughout the lease term. Certain of these leases also include renewal options at the election of the Company to renew or extend the lease for additional years. These optional periods have not been considered in the determination of the ROU or lease liabilities associated with these leases as management did not consider it reasonably certain it would exercise the options. Short-term leases, which have an initial term of 12 months or less, are not recorded in the balance sheet and expense for these leases is recognized on a straight-line basis over the lease term.
The Company’s lease agreements do not contain any termination options, material residual value guarantees, material bargain purchase options or material restrictive covenants. The Company does not have any lease transactions with related parties.
Total lease cost includes the following components for the years ended December 31, 2021 and 2020:
| December 31, | ||||||||||||||
| 2021 | 2020 | |||||||||||||
| (in thousands) | ||||||||||||||
| Operating lease expense cost | $ | 656 | $ | 644 | ||||||||||
Finance Lease Costs | ||||||||||||||
| Interest expense | 7 | 24 | ||||||||||||
| Amortization expense | 126 | 80 | ||||||||||||
| Total finance lease costs | $ | 133 | $ | 104 | ||||||||||
F-25
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
| December 31, | ||||||||||||||
| 2021 | 2020 | |||||||||||||
Supplemental balance sheet information | (in thousands) | |||||||||||||
Operating leases | ||||||||||||||
| Operating lease right-of-use assets | $ | 2,206 | $ | 2,610 | ||||||||||
| Operating lease liabilities - short-term | 402 | 788 | ||||||||||||
| Operating lease liabilities - long-term | 1,900 | 1,923 | ||||||||||||
| Total operating lease liabilities | $ | 2,302 | $ | 2,711 | ||||||||||
Finance leases | ||||||||||||||
| Finance lease right-of-use assets | $ | 154 | $ | 313 | ||||||||||
| Finance lease liabilities - short-term | 13 | 160 | ||||||||||||
| Finance lease liabilities - long-term | — | 28 | ||||||||||||
| Total finance lease liabilities | $ | 13 | $ | 188 | ||||||||||
| Weighted-average remaining lease term (years) | ||||||||||||||
| Operating leases | 5.5 | 6.2 | ||||||||||||
| Finance leases | 0.8 | 1.0 | ||||||||||||
Weighted-average discount rate (%) | ||||||||||||||
| Operating leases | 10.4 | % | 10.5 | % | ||||||||||
| Finance leases | 8.3 | % | 9.1 | % | ||||||||||
F-26
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
| December 31, | ||||||||||||||
| 2021 | 2020 | |||||||||||||
| Cash paid for amounts included in the measurement of lease liabilities | (in thousands) | |||||||||||||
| Operating cash outflows from operating leases | $ | 639 | $ | 137 | ||||||||||
| Operating cash outflows from finance leases | $ | 7 | $ | 24 | ||||||||||
| Financing cash outflows from finance leases | $ | 175 | $ | 277 | ||||||||||
| ROU assets obtained in exchange for new lease liabilities | ||||||||||||||
| Operating leases | $ | — | $ | 355 | ||||||||||
| Finance leases | $ | — | $ | — | ||||||||||
Maturities of lease liabilities as of December 31, 2021 were as follows:
| Years Ending December 31, | Operating Leases | Finance Leases | ||||||||||||
| (in thousands) | ||||||||||||||
| 2022 | $ | 563 | $ | 14 | ||||||||||
| 2023 | 535 | — | ||||||||||||
| 2024 | 504 | — | ||||||||||||
| 2025 | 429 | — | ||||||||||||
| 2026 | 404 | — | ||||||||||||
| Thereafter | 436 | — | ||||||||||||
| Total future minimum lease payments | 2,871 | 14 | ||||||||||||
| Less: Amount of lease payments representing interest | (569) | (1) | ||||||||||||
| Present value of future minimum lease payments | $ | 2,302 | $ | 13 | ||||||||||
8. Shareholders’ Equity
Under the Memorandum of Association and Articles of Association, or Articles, in effect as of December 31, 2021 and 2020, the Company had authorized an unlimited number of common shares with no par value.
As of December 31, 2021 and 2020, 24,488,335 and 23,925,789 common shares, respectively, were issued and 24,080,265 and 23,517,719 common shares, respectively, were outstanding.
During the year ended December 31, 2021, the Company granted stock options to employees and contractors (see Note 10).
F-27
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
The Company had reserved common shares for future issuances at December 31:
| 2021 | 2020 | |||||||||||||
| Warrants to purchase common shares | 5,500 | 5,500 | ||||||||||||
| Options to purchase common shares | 2,098,087 | 2,012,960 | ||||||||||||
| Remaining shares available under the 2018 Equity Incentive Plan | 1,780,687 | 1,641,112 | ||||||||||||
| Shares issuable on vesting of grants of restricted stock | 3,982 | 48,624 | ||||||||||||
| Remaining shares available under the 2018 ESPP | 661,000 | 474,000 | ||||||||||||
| Total | 4,549,256 | 4,182,196 | ||||||||||||
9. Warrants
In March 2017, the Company issued warrants for the purchase of 145,000 Class B ordinary shares to parties related to Rockport Ventures, with a fixed exercise price of $3.80 per share.
During the year ended December 31, 2021, no warrants were exercised. As of December 31, 2021 and 2020, 5,500 warrants to purchase the Company’s common shares were outstanding and exercisable:
| Warrant Holder | Issue Date | In Connection With | Warrant to Purchase | Shares | Exercise Price | Expiration Date | ||||||||||||||||||||||||||||||||
| Rockport | 3/3/2017 | Loan agreement | Common | 5,500 | $ | 3.80 | 8/28/2022 | |||||||||||||||||||||||||||||||
10. Share-Based Compensation
In 2015, the Board of Directors approved and adopted the 2015 Equity Incentive Plan, or 2015 Plan. Pursuant to the 2015 Plan, the Company granted RSAs and stock options to members of the Board of Directors, employees and consultants.
In 2018, the Board of Directors terminated the 2015 Plan and approved the 2018 Equity Incentive Plan, or the 2018 Plan, with an initial reserve of 1,500,000 common shares. Under the 2018 Plan, the Company may grant stock options, equity appreciation rights, and restricted share awards . If an award granted under the 2018 Plan expires, terminates, is unexercised, or is forfeited, or if any shares are surrendered in connection with an incentive award, the shares subject to such award and the surrendered shares become available for further awards under the 2018 Plan.
Pursuant to the “evergreen” provision contained in the 2018 Plan, the number of common shares reserved for issuance under the 2018 Plan automatically increases on first day of each fiscal year, commencing on January 1, 2019, in an amount equal to the least of (1) 750,000 shares, (2) 4% of the total number of the Company’s common shares outstanding on the last day of the preceding fiscal year, or (3) a number of common shares as may be determined by the Company’s Board of Directors prior to any such increase date. On each of January 1, 2019, 2020, and 2021 the number of common shares authorized for issuance increased automatically by 750,000 shares in accordance with the evergreen provision, increasing the number of common shares reserved under the 2018 Plan to 3,750,000 as of December 31, 2021.
During the periods presented, the Company recorded the following share-based compensation expense for stock
F-28
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
options and restricted stock awards:
| Year Ended December 31, | ||||||||||||||||||||
| 2021 | 2020 | 2019 | ||||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Sales, general and administrative | $ | 7,908 | $ | 4,203 | $ | 5,021 | ||||||||||||||
| Research and development | 2,499 | 1,518 | 1,505 | |||||||||||||||||
| Total | $ | 10,407 | $ | 5,721 | $ | 6,526 | ||||||||||||||
Stock Options
| Number of Options Outstanding | Weighted-Average Exercise Price | Weighted-Average Remaining Contractual Term (in years) | Aggregate Intrinsic Value (in thousands) | |||||||||||||||||||||||
| Balances at December 31, 2020 | 2,012,960 | $ | 16.71 | 7.75 | $ | 42,126 | ||||||||||||||||||||
Granted (weighted-average fair value of $40.98 per share) | 775,003 | 73.00 | ||||||||||||||||||||||||
| Exercised | (521,316) | 11.94 | ||||||||||||||||||||||||
| Forfeited/canceled | (168,560) | 31.67 | ||||||||||||||||||||||||
| Balances at December 31, 2021 | 2,098,087 | $ | 37.49 | 7.67 | $ | 67,357 | ||||||||||||||||||||
As of December 31, 2021, 850,727 options were vested and exercisable with weighted-average exercise price of $15.63 per share and a total aggregate intrinsic value of $44.2 million.
During the year ended December 31, 2021, 521,316 options were exercised at a weighted-average price of $11.94 per share. The intrinsic value of the options exercised during the years ended December 31, 2021, 2020, and 2019 was $28.7 million, $1.9 million, and $1.5 million, respectively. Upon the exercise of stock options, the Company issued new shares from its authorized shares.
At December 31, 2021, unrecognized compensation expense was $27.7 million related to stock options granted to employees and members of the Board of Directors and $1.3 million related to stock options granted to consultants. The weighted-average period over which such compensation expense will be recognized is 2.6 years.
Stock Options Granted to Employees
Share-based compensation expense for employees is based on the grant date fair value. The Company recognizes compensation expense for all share-based awards ratably on a straight-line basis over the requisite service period of the awards, which is generally the vesting term of four years. During the year ended December 31, 2021, 2020 and 2019, the Company recognized $8.2 million, $2.8 million and $2.2 million, respectively, of share-based compensation expense for stock options granted to employees.
The Company uses the Black-Scholes option valuation model to value options granted to employees and consultants, which requires the use of highly subjective assumptions to determine the fair value of share-based awards. The assumptions used in the Company’s option-pricing model represent management’s best estimates. These estimates are complex, involve a number of variables, uncertainties and assumptions and the application of management’s judgment. If factors change and different assumptions are used, the Company’s share-based
F-29
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
compensation expense could be materially different in the future. The assumptions and estimates that the Company uses in the Black-Scholes model are as follows:
▪Fair Value of Common Shares. Following the Company’s IPO in 2018, the closing price of the Company’s publicly-traded common shares on the date of grant is used as the fair value of the shares. Prior to the IPO, the fair value of ordinary shares was estimated on a periodic basis by the Company’s Board of Directors, with the assistance of an independent third-party valuation firm. The Board of Directors intended all options granted to be exercisable at a price per share not less than the estimated per share fair value of the shares underlying those options on the date of grant.
▪Risk-Free Interest Rate. The Company bases the risk-free interest rate used in the Black-Scholes valuation model on the implied yield available on U.S. Treasury zero-coupon issues with a term equivalent to that of the term of the options for each option group on the measurement date.
▪Term. For employee stock options, the expected term represents the period that the Company’s share-based awards are expected to be outstanding. Because of the limitations on the sale or transfer of the Company’s shares during the period the Company was a privately held company, the Company does not believe its historical exercise pattern is indicative of the pattern it will experience as a publicly traded company. The Company consequently uses the Staff Accounting Bulletin 110, or SAB 110, simplified method to calculate the expected term of employee stock options, which is the average of the contractual term and vesting period. The Company plans to continue to use the SAB 110 simplified method until it has sufficient trading history as a publicly traded company. For consultant stock options, the term used is equal to the remaining contractual term on the measurement date.
▪Volatility. The Company determines the price volatility based on the historical volatilities of industry peers as it does not have sufficient trading history for its shares. Industry peers consist of several public companies in the medical device industry with comparable characteristics, including revenue growth, operating model and working capital requirements. The Company intends to continue to consistently apply this process using the same or a similar peer group of public companies until a sufficient amount of historical information regarding the volatility of its own shares becomes available, or unless circumstances change such that the identified peer companies are no longer similar, in which case other suitable peer companies whose common share prices are publicly available would be utilized in the calculation. The volatility is calculated based on the term on the measurement date.
▪Dividend Yield. The expected dividend assumption is based on the Company’s current expectations about its anticipated dividend policy. The Company has no expectation that it will declare dividends on its common shares, and therefore has used an expected dividend yield of zero.
The fair value of stock options granted to employees was estimated using the following assumptions:
| Year Ended December 31, | ||||||||||||||||||||
| 2021 | 2020 | 2019 | ||||||||||||||||||
| Volatility | 60% | 55% - 60% | 56% | |||||||||||||||||
| Risk-free interest rate | 0.7% - 1.4% | 0.4% - 1.5% | 1.6% - 2.6% | |||||||||||||||||
| Term (in years) | 6.25 | 6.25 | 6.25 | |||||||||||||||||
| Dividend yield | — | — | — | |||||||||||||||||
Stock Options Granted to Non-Employees
Share-based compensation expense related to stock options granted to non-employees is recognized as the stock options are earned using an accelerated attribution method. The Company believes that the estimated fair value of the stock options is more readily measurable than the fair value of the services rendered. For the years ended December 31, 2021, 2020 and 2019, the Company recognized expense of $1.8 million, $2.3 million and $3.2 million, respectively, for stock options granted to consultants.
F-30
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
The fair value of stock options granted to consultants was estimated using the following assumptions during the following periods presented:
| Year Ended December 31, | ||||||||||||||||||||
| 2021 | 2020 | 2019 | ||||||||||||||||||
| Volatility | 60% | 56% - 60% | 56% - 57% | |||||||||||||||||
| Risk-free interest rate | 1.6% | 0.6% - 1.6% | 1.7% - 2.1% | |||||||||||||||||
| Term (in years) | 10 | 10 | 10 | |||||||||||||||||
| Dividend yield | — | — | — | |||||||||||||||||
Restricted Stock
Each vested RSA or RSU entitles the holder to be issued one common share. These awards vest according to a vesting schedule determined by the Compensation Committee of the Company’s Board of Directors, generally over a to four year period.
The following table represents restricted stock activity for fiscal 2021:
| Restricted Stock | Weighted- Average Grant Date Fair Value | |||||||||||||
| Outstanding unvested at December 31, 2020 | 48,624 | $ | 11.32 | |||||||||||
| Granted | 3,982 | 69.05 | ||||||||||||
| Vested | (48,124) | 11.34 | ||||||||||||
| Forfeited/canceled | (500) | 9.64 | ||||||||||||
| Outstanding unvested at December 31, 2021 | 3,982 | $ | 69.05 | |||||||||||
The fair value of restricted stock is the grant date market value of common shares. The Company recognizes share-based compensation expense related to restricted stock using a straight-line method over the vesting term of the awards. The share-based compensation expense for restricted stock that vested during the years ended December 31, 2021, 2020 and 2019 was $0.4 million, $0.6 million and $1.1 million, respectively, which was calculated based on the market value of the Company’s common shares on the applicable grant date.
As of December 31, 2021, we had unrecognized share-based compensation cost of approximately $0.3 million associated with unvested awards of restricted stock. This cost is expected to be recognized over a weighted-average period of approximately 1.0 year.
F-31
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
11. Income Taxes
For the year ended December 31, loss before income tax consisted of the following:
| 2021 | 2020 | 2019 | |||||||||||||||
| (in thousands) | |||||||||||||||||
| Costa Rica operations | $ | 4,027 | $ | (8,872) | $ | 5,022 | |||||||||||
| Non-Costa Rica operations | (43,739) | (29,145) | (42,532) | ||||||||||||||
| $ | (39,712) | $ | (38,017) | $ | (37,510) | ||||||||||||
For the year ended December 31, the income tax provision (benefit) consisted of the following:
| 2021 | 2020 | 2019 | |||||||||||||||
| (in thousands) | |||||||||||||||||
| Current | |||||||||||||||||
| Costa Rica | $ | 289 | $ | — | $ | — | |||||||||||
| Non-Costa Rica | 1,131 | 378 | 640 | ||||||||||||||
| Total current | 1,420 | 378 | 640 | ||||||||||||||
| Deferred | |||||||||||||||||
| Costa Rica | — | — | — | ||||||||||||||
| Non-Costa Rica | 7 | (274) | — | ||||||||||||||
| Total deferred | 7 | (274) | — | ||||||||||||||
| Total provision | $ | 1,427 | $ | 104 | $ | 640 | |||||||||||
F-32
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
The items accounting for the difference between income taxes computed at the Costa Rica statutory income tax rate and the income tax provision (benefit) consisted of the following for the year ended December 31:
| 2021 | 2020 | 2019 | |||||||||||||||||||||||||||||||||
| (in thousands) | |||||||||||||||||||||||||||||||||||
| Tax benefit at Costa Rica statutory rate | $ | (11,914) | 30 | % | $ | (11,405) | 30 | % | $ | (11,253) | 30 | % | |||||||||||||||||||||||
| Foreign tax rate differential | 9,430 | (24) | 5,252 | (14) | 10,623 | (29) | |||||||||||||||||||||||||||||
| Return to provision adjustment | 384 | (1) | (559) | 2 | 391 | (1) | |||||||||||||||||||||||||||||
| Tax credits | (53) | — | (82) | — | (56) | — | |||||||||||||||||||||||||||||
| Change in valuation allowance | 7,043 | (18) | 4,071 | (11) | 2,271 | (6) | |||||||||||||||||||||||||||||
| Tax holiday adjustment (benefit) | (918) | 2 | 2,636 | (7) | (1,507) | 4 | |||||||||||||||||||||||||||||
| U.S. Stock Compensation | (2,681) | 7 | — | — | — | — | |||||||||||||||||||||||||||||
| Other | 136 | — | 191 | — | 171 | — | |||||||||||||||||||||||||||||
| Total provision for income taxes | $ | 1,427 | (4) | % | $ | 104 | — | % | $ | 640 | (2) | % | |||||||||||||||||||||||
The Company's tax holiday benefit was related to the Company’s subsidiary in Costa Rica which enjoyed a zero percent tax rate, with the exception of extended warranty income, for the years ended December 31, 2021, 2020 and 2019. The zero percent tax holiday was granted in August 2018 for a period of 8 years through the year 2026.
As of December 31, the components of the Company's deferred tax assets and liabilities are as follows:
| 2021 | 2020 | ||||||||||
| (in thousands) | |||||||||||
| Accruals and reserves | $ | 301 | $ | 133 | |||||||
| Intangibles | 130 | 113 | |||||||||
| Stock compensation | 775 | 197 | |||||||||
| Net operating loss | 18,208 | 11,868 | |||||||||
| R&D credits | 169 | 116 | |||||||||
| Other | 44 | 179 | |||||||||
| Valuation allowance | (19,369) | (12,332) | |||||||||
| Total net deferred tax assets (included in “Other non-current assets”) | $ | 258 | $ | 274 | |||||||
As of December 31, 2021, the Company assessed that it is more-likely-than-not that it will not realize its deferred tax assets based on the absence of sufficient positive objective evidence that it would generate sufficient taxable income in its Brazil and U.S. tax jurisdiction (Motiva USA, LLC) to realize the deferred tax assets. The Company intends to continue maintaining a full valuation allowance on its deferred tax assets in these jurisdictions until there is sufficient evidence to support the reversal of all or some portion of these allowances.
As of December 31, 2021, the Company has U.S. and California tax credit carryforwards of approximately $0.2 million in total. The federal research credits begin to expire in 2037. However, the California research credits can be carried forward indefinitely.
F-33
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
As of December 31, 2021, the Company had U.S. federal, state, U.K. and Brazil net operating losses of approximately $52.6 million, $6.8 million, $0.1 million and $20.3 million, respectively. The U.S. federal net operating losses of $3.3 million generated prior to 2018, and state net operating losses will begin to expire on December 31, 2030. The U.S. federal net operating losses generated in 2018 and future years will be carried forward indefinitely. Brazil net operating losses can be carried forward indefinitely.
The United States federal and California laws impose restrictions on the utilization of net operating loss carryforwards and R&D credit carryforwards in the event of a change in ownership of the Company, which constitutes an “ownership change” as defined by Internal Revenue Code Sections 382 and 383. Generally, an ownership change occurs if the percentage of the value of the shares that are owned by one or more direct or indirect “five percent shareholders” increases by more than 50% over their lowest ownership percentage at any time during the applicable testing period. If the Company has experienced an “ownership change” at any time since its formation, it would already be subject to limitations on its ability to utilize its existing net operating losses and other tax attributes. The Company did not experience an ownership change in the past that would materially impact the availability of its net operating losses and tax credits. Nevertheless, future changes in our share ownership, which may be outside of the Company’s control, may trigger an “ownership change” and, consequently, Section 382 and 383 limitations. The Company has not completed a Section 382 and 383 analysis to determine if an ownership change has occurred. Until such analysis is completed, the Company cannot be sure that the full amount of the existing net operating loss carryforwards will be available, even if the Company does generate taxable income before their expiration. In addition, under the newly enacted U.S. federal income tax law, federal net operating losses incurred in 2018 and in future years may be carried forward indefinitely, but the deductibility of such federal net operating losses is limited.
Discontinuation of preferential tax treatment the Company currently enjoys or other unfavorable changes in tax law could result in additional compliance obligations and costs. The Company is currently the beneficiary of a tax holiday in Costa Rica pursuant to which it is subject to a tax at a 0% rate, with the exception of the extended warranty sales income that is subject to 30% income tax. However, there can be no assurance that the Company will continue to qualify for or receive such favorable tax treatment. If the Company fails to maintain such favorable tax treatment it may be subject to tax in Costa Rica at a significantly higher rate.
A tax authority may disagree with tax positions that the Company has taken, which could result in increased tax liabilities. For example, the U.S. Internal Revenue Service or another tax authority could challenge the amounts paid between the Company and its subsidiaries pursuant to the Company’s intercompany arrangements and transfer pricing policies. A tax authority may take the position that material income tax liabilities, interest and penalties are payable by the Company, in which case, the Company expects that it might contest such assessment. Contesting such an assessment may be lengthy and costly and, if the Company is unsuccessful in disputing the assessment, the implications could increase its anticipated effective tax rate, where applicable. In addition, the Company may be subject to additional tax liabilities, which could materially and adversely affect its business, financial condition and results of operations. The application, interpretation and enforcement of the value-added tax, or VAT, and other taxes and related regulations applicable to medical device companies are complex and evolving.
The Company conducts operations in multiple jurisdictions and is subject to certain taxes, including income, sales and use, employment, value added and other taxes, in the United States and other jurisdictions in which the Company does business. A change in the tax laws in the jurisdictions in which the Company does business, including an increase in tax rates or an adverse change in the treatment of an item of income or expense, possibly with retroactive effect, could result in a material increase in the amount of taxes incurred.
The Company’s determination of its tax liability is subject to review by applicable U.S. and foreign tax authorities. Any adverse outcome of such a review could harm the Company’s operating results and financial condition. The determination of the Company’s worldwide provision for income taxes and other tax liabilities requires significant judgment and, in the ordinary course of business, there are many transactions and calculations where the ultimate tax determination is complex and uncertain. Moreover, as a multinational business, the Company has subsidiaries that engage in many intercompany transactions in a variety of tax jurisdictions where the ultimate tax determination is complex and uncertain. The taxing authorities of the jurisdictions in which the Company operates may challenge the Company’s methodologies, which could impact its financial position and operating results.
F-34
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
Historically, the Company has allocated some of its employees’ and contractors’ time across multiple business entities in the international jurisdictions in which the Company operates. If the Company determined that it had misclassified the employees’ or contractors’ employment status or certain of its expenditures under local laws, the Company may be subjected to penalties or be required to pay withholding taxes for, extend employee benefits to, provide compensation for unpaid overtime to, or otherwise incur substantially greater expenses with respect to such employees and contractors. Any of the foregoing circumstances could have a material adverse impact on the Company’s operating results and financial condition.
The Company is periodically reviewed and audited by tax authorities with respect to income and non-income taxes. Tax authorities may disagree with certain positions the Company has taken, and we may have exposure to additional income and non-income tax liabilities which could have an adverse effect on the Company’s operating results and financial condition. Such authorities could impose additional taxes, interest and penalties, claim that various withholding requirements apply to the Company or its subsidiaries or assert that benefits of tax treaties are not available to the Company or its subsidiaries. In addition, the Company’s future effective tax rates could be favorably or unfavorably affected by changes in tax rates, changes in the valuation of the Company’s deferred tax assets or liabilities, the effectiveness of its tax planning strategies, or changes in tax laws or their interpretation. Such changes could have an adverse impact on the Company’s financial condition.
As a result of these and other factors, the ultimate amount of tax obligations may differ from the amounts recorded in the Company’s financial statements and any such difference may harm its operating results in future periods in which the Company changes the estimates of such tax obligations or in which the ultimate tax outcome is determined.
A non-U.S. corporation is classified as a passive foreign investment company, or PFIC, for U.S. federal income tax purposes, in any taxable year in which either (1) at least 75% of its gross income is passive income; or (2) at least 50% of the average quarterly value of its total gross assets is attributable to assets that produce “passive income” or are held for the production of passive income. Based on the project composition of the Company’s income and valuation of its assets, the Company does not believe it is a PFIC in 2021, and the Company does not expect to be a PFIC for the current taxable year or to become one in the future. However, because the PFIC status is subject to a number of uncertainties, neither the Company nor its tax advisors can provide any assurances regarding the PFIC status. If the Company is a PFIC for any taxable year during which a U.S. holder holds the Company’s common shares, the U.S. holder may be subject to adverse tax consequences.
The Company records income taxes using the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in the consolidated financial statements or income tax returns. In estimating future tax consequences, expected future events, enactments or changes in the tax law or rates are considered. Valuation allowances are provided when necessary to reduce deferred tax assets to the amount expected to be realized.
Accounting for Uncertainty in Income Taxes
The Company records uncertain tax positions based on a two-step process whereby (1) a determination is made as to whether it is more likely than not that the tax positions will be sustained based on the technical merits of the position and (2) for those tax positions that meet the more-likely-than-not recognition threshold the Company recognizes the largest amount of tax benefit that is greater than 50% likely to be realized upon ultimate settlement with the related tax authority. The Company’s policy is to recognize interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense. Significant judgment is required in the identification of uncertain tax positions and in the estimation of penalties and interest on uncertain tax positions.
The Company has adopted ASC 740-10 Accounting for Uncertainty in Income Taxes (formerly FIN 48). ASC 740-10 prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of uncertain tax positions taken or expected to be taken in the Company's income tax return, and provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure, and transition. For the years ended December 31, 2021 and 2020 the Company has no material uncertain tax positions. The Company has R&D credits in the United States and has recorded reserves of $28,000 which offsets R&D credit deferred tax assets. The Company does not expect any significant increases or
F-35
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
decreases to its unrecognized tax benefits within the next 12 months. The Company’s policy is to recognize interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense.
As of December 31, 2021, the Company is subject to taxation in Costa Rica, Belgium, France, Brazil, the United Kingdom, Sweden, Italy, Germany, Austria, Spain, Argentina, Switzerland and the United States and the Company’s fiscal tax years 2017 through 2021 are subject to examination by the tax authorities.
12. Net Loss Per Share
The following table summarizes the computation of basic and diluted net loss per share for the periods presented:
| Year Ended December 31, | ||||||||||||||||||||
| 2021 | 2020 | 2019 | ||||||||||||||||||
| (in thousands, except share and per share data) | ||||||||||||||||||||
| Numerator: | ||||||||||||||||||||
| Net loss | $ | (41,139) | $ | (38,121) | $ | (38,150) | ||||||||||||||
| Denominator: | ||||||||||||||||||||
| Weighted average common shares used for basic and diluted earnings per share | 23,972,722 | 23,316,102 | 20,541,528 | |||||||||||||||||
| Net loss per share: | ||||||||||||||||||||
Basic and diluted | $ | (1.72) | $ | (1.63) | $ | (1.86) | ||||||||||||||
Basic net loss per share is computed by dividing the net loss by the weighted-average number of shares outstanding for the period. Diluted net loss per share is computed by dividing the net loss by the weighted-average number of shares and dilutive share equivalents outstanding for the period, determined using the treasury-share method and the as-if converted method, for convertible securities, if inclusion of these is dilutive.
If the Company reports a net loss, diluted net loss per share is the same as basic net loss per share for those periods because including the dilutive securities would be anti-dilutive.
The following potentially dilutive securities outstanding at the end of the periods presented have been excluded from the computation of diluted shares:
| Year Ended December 31, | ||||||||||||||||||||
| 2021 | 2020 | 2019 | ||||||||||||||||||
| Options to purchase common shares | 1,873,087 | 1,752,620 | 1,689,016 | |||||||||||||||||
| Shares issuable on vesting of grants of restricted stock | 3,982 | 48,624 | 128,682 | |||||||||||||||||
| Warrants to purchase common shares | 5,500 | 5,500 | 5,500 | |||||||||||||||||
| Total | 1,882,569 | 1,806,744 | 1,823,198 | |||||||||||||||||
13. Related Party Transactions
During the years ended December 31, 2021, 2020 and 2019, the Company recorded revenue of $1.4 million, $0.9 million and $0.7 million, respectively, for product sales to Herramientas Medicas, S.A., a distribution company owned by a family member of the Chief Executive Officer of the Company. Accounts receivable owed to the Company from this distribution company amounted to approximately $0.4 million and $0.2 million as of December 31, 2021 and 2020, respectively.
F-36
ESTABLISHMENT LABS HOLDINGS, INC.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2021, 2020 and 2019
In 2016, the Company also entered into a separate agreement with Dr. Chacón Quirós, the brother of the Company’s Chief Executive Officer Juan José Chacón Quirós, to maintain his clinic in Costa Rica as a MotivaImagine Excellence Center and to host and train physicians in the use of the Company products in relevant procedures, among other services, in exchange for cash reimbursement of up to $4,500 per day that such services are rendered. In December 2020, Dr. Chacón Quirós was granted options to purchase 22,068 shares vesting over four years in equal annual installments, provided that he continues to provide these services at such times. During the years ended December 31, 2021, 2020 and 2019, the Company paid Dr. Chacón Quirós approximately $0.4 million, $0.1 million and $0.1 million, respectively, for services rendered.
14. Employee Benefits
Short-term employee benefits, including vacation (paid absences) and year-end bonuses (also known as 13th month salary), are current liabilities included in accrued liabilities on the consolidated balance sheets and are calculated at the non-discounted amount that the Company expects to pay as a result of uncharged employee salaries or retentions.
Regarding employee termination benefits, Costa Rica labor laws establish the payment of benefits in case of death, retirement or termination without cause. This compensation is calculated according to time served in the Company and the corresponding salary in the last six months of employment and is equal to between 19.5 and 22 days salary for each year served, up to a maximum of 8 years.
Company policy recognizes termination benefits as expenses of the period during which the termination occurs, when the legal obligation is assumed due to the aforementioned events.
The 47 employees in Brazil are represented by a labor union.
15. Commitments and Contingencies
Contingencies
Periodically, the Company may have certain contingent liabilities that arise in the ordinary course of business activities. The Company accrues a liability for such matters when it is probable that future expenditures will be made, and such expenditures can be reasonably estimated. There have been no contingent liabilities requiring accrual or disclosure at December 31, 2021 and 2020.
Indemnification
The Company enters into standard indemnification arrangements in the ordinary course of business. Pursuant to these arrangements, the Company indemnifies, holds harmless, and agrees to reimburse the indemnified parties for losses suffered or incurred by the indemnified party, in connection with any trade secret, copyright, patent or other intellectual property infringement claim by any third party with respect to the Company’s technology. The term of these indemnification agreements is generally perpetual. The maximum potential amount of future payments the Company could be required to make under these agreements is not determinable because it involves claims that may be made against the Company in the future, but have not yet been made.
The Company has entered into indemnification agreements with its directors and officers that may require the Company to indemnify its directors and officers against liabilities that may arise by reason of their status or service as directors or officers, other than liabilities arising from willful misconduct of the individual.
The Company has not incurred costs to defend lawsuits or settle claims related to these indemnification agreements. No liability associated with such indemnifications has been recorded to date.
F-37
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
ESTABLISHMENT LABS HOLDINGS INC. | |||||||||||
| Dated: | March 1, 2022 | By: | /s/ Juan José Chacón Quirós | ||||||||
| Name: | Juan José Chacón Quirós | ||||||||||
| Title: | Chief Executive Officer and Director | ||||||||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||||||||||||
/s/ Juan José Chacón Quirós | Chief Executive Officer and Director (Principal Executive Officer) | March 1, 2022 | ||||||||||||
Juan José Chacón Quirós | ||||||||||||||
/s/ Rajbir S. Denhoy | Chief Financial Officer (Principal Financial and Accounting Officer) (Authorized Representative in the United States) | March 1, 2022 | ||||||||||||
| Rajbir S. Denhoy | ||||||||||||||
| /s/ Nicholas Lewin | Chairman of the Board of Directors | March 1, 2022 | ||||||||||||
| Nicholas Lewin | ||||||||||||||
| /s/ Lisa N. Colleran | Director | March 1, 2022 | ||||||||||||
| Lisa N. Colleran | ||||||||||||||
| /s/ Dennis Condon | Director | March 1, 2022 | ||||||||||||
| Dennis Condon | ||||||||||||||
| /s/ Ann Custin | Director | March 1, 2022 | ||||||||||||
| Ann Custin | ||||||||||||||
| /s/ Lisa Gersh | Director | March 1, 2022 | ||||||||||||
| Lisa Gersh | ||||||||||||||
| /s/ Leslie Gillin | Director | March 1, 2022 | ||||||||||||
| Leslie Gillin | ||||||||||||||
| /s/ Edward Schutter | Director | March 1, 2022 | ||||||||||||
| Edward Schutter | ||||||||||||||
| /s/ Bryan Slotkin | Director | March 1, 2022 | ||||||||||||
| Bryan Slotkin | ||||||||||||||
F-38
Similar companies
See also INTUITIVE SURGICAL INC - Annual report 2022 (10-K 2022-12-31) Annual report 2023 (10-Q 2023-09-30)See also Edwards Lifesciences Corp - Annual report 2022 (10-K 2022-12-31) Annual report 2023 (10-Q 2023-09-30)
See also ZIMMER BIOMET HOLDINGS, INC. - Annual report 2022 (10-K 2022-12-31) Annual report 2023 (10-Q 2023-09-30)
See also STERIS plc - Annual report 2023 (10-K 2023-03-31) Annual report 2023 (10-Q 2023-09-30)
See also ALIGN TECHNOLOGY INC - Annual report 2022 (10-K 2022-12-31) Annual report 2023 (10-Q 2023-09-30)