MARINUS PHARMACEUTICALS, INC. - Quarter Report: 2022 March (Form 10-Q)
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-Q
☒ | QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE QUARTERLY PERIOD ENDED MARCH 31, 2022
OR
☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
COMMISSION FILE NUMBER 001-36576

MARINUS PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
Delaware | 20-0198082 | |
(State or other jurisdiction of | (I.R.S. Employer |
5 Radnor Corporate Center, Suite 500
100 Matsonford Rd
Radnor, PA 19087
(Address of registrant’s principal executive offices, including zip code)
Registrant’s telephone number, including area code: (484) 801-4670
Securities registered pursuant to Section 12(b) of the Act:
Title of Each Class | Trading Symbol(s) | Name of Each Exchange on Which Registered | ||
Common Stock, par value $0.001 per share | MRNS | Nasdaq Global Market |
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ⌧ Yes☐ No.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). ☒ Yes ☐ No.
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ☐ | Accelerated filer | ☐ | |
Non-accelerated filer ☒ | Smaller reporting company | ☒ | |
Emerging growth company ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ◻
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). ☐Yes ☒ No.
The number of outstanding shares of the registrant’s common stock, par value $0.001 per share, as of April 30, 2022, was: 37,140,406.
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
INDEX TO FORM 10-Q
FOR THE QUARTER ENDED MARCH 31, 2022
Unless the context otherwise requires, all references in this Quarterly Report on Form 10-Q to the “Company,” “Marinus,” “we,” “us,” and “our” include Marinus Pharmaceuticals, Inc. and its wholly owned subsidiary, Marinus Pharmaceuticals Emerald Limited, an Ireland company.
2
PART I
FINANCIAL INFORMATION
Item 1. Consolidated Financial Statements
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED BALANCE SHEETS
(in thousands, except share and per share amounts)
(unaudited)
March 31, | December 31, | ||||||
2022 | 2021 | ||||||
ASSETS |
|
|
|
| |||
Current assets: | |||||||
Cash and cash equivalents | $ | 126,319 | $ | 122,927 | |||
Accounts receivable | 2,489 | 2,629 | |||||
Contract asset | — | 557 | |||||
Prepaid expenses and other current assets |
| 6,263 |
| 5,565 | |||
Total current assets |
| 135,071 |
| 131,678 | |||
Property and equipment, net |
| 2,989 |
| 2,499 | |||
Other assets |
| 2,750 |
| 2,663 | |||
Total assets | $ | 140,810 | $ | 136,840 | |||
LIABILITIES AND STOCKHOLDERS’ EQUITY | |||||||
Current liabilities: | |||||||
Accounts payable | $ | 4,671 | $ | 3,126 | |||
Refund liability |
| — |
| 21,233 | |||
Accrued expenses | 14,865 | 16,207 | |||||
Total current liabilities |
| 19,536 |
| 40,566 | |||
Notes payable, net of deferred financing costs | 69,927 | 40,809 | |||||
Contract liability | 9,113 | — | |||||
Other long-term liabilities | 1,829 | 1,979 | |||||
Total liabilities | 100,405 | 83,354 | |||||
Stockholders’ equity: | |||||||
Series A convertible preferred stock, $0.001 par value; 25,000,000 shares authorized, 4,575 shares issued and outstanding at March 31, 2022 and December 31, 2021 | 4,302 | 4,302 | |||||
Common stock, $0.001 par value; 150,000,000 shares authorized, 37,145,981 issued and 37,138,674 outstanding at March 31, 2022 and 36,797,561 issued and 36,790,254 outstanding at December 31, 2021 |
| 37 |
| 37 | |||
Additional paid-in capital |
| 466,132 |
| 459,852 | |||
Treasury stock at cost, 7,307 shares at March 31, 2022 and December 31, 2021 |
|
| |||||
Accumulated deficit |
| (430,066) |
| (410,705) | |||
Total stockholders’ equity |
| 40,405 |
| 53,486 | |||
Total liabilities and stockholders’ equity | $ | 140,810 | $ | 136,840 | |||
See accompanying notes to consolidated financial statements.
3
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(in thousands, except share and per share amounts)
(unaudited)
Three Months Ended March 31, | ||||||||
| 2022 |
| 2021 |
| ||||
Revenue: |
|
| ||||||
Federal contract revenue |
| $ | 1,513 |
| $ | 1,806 |
| |
Collaboration revenue |
| 12,673 |
| — |
| |||
Total revenue | 14,186 | 1,806 | ||||||
Expenses: | ||||||||
Research and development | 17,991 | 18,591 | ||||||
General and administrative |
| 11,737 |
| 10,376 | ||||
Cost of IP license fee |
| 1,169 |
| — | ||||
Total expenses |
| 30,897 |
| 28,967 | ||||
Loss from operations |
| (16,711) |
| (27,161) | ||||
Interest income |
| 12 |
| 24 | ||||
Interest expense |
| (1,692) |
| — | ||||
Other expense, net |
| (970) |
| (4) | ||||
Net loss and comprehensive loss | $ | (19,361) | $ | (27,141) | ||||
Net loss applicable to common shareholders | $ | (19,361) | $ | (27,141) | ||||
Per share information: | ||||||||
Net loss per share of common stock—basic and diluted | (0.52) | (0.74) | ||||||
Basic and diluted weighted average shares outstanding |
| 36,890,568 |
| 36,599,701 | ||||
See accompanying notes to consolidated financial statements.
4
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
(unaudited)
Three Months Ended March 31, |
| ||||||
2022 | 2021 |
| |||||
| |||||||
Cash flows from operating activities |
|
|
|
| |||
Net loss | $ | (19,361) | $ | (27,141) | |||
Adjustments to reconcile net loss to net cash used in operating activities: | |||||||
Depreciation and amortization |
| 85 |
| 85 | |||
Amortization of debt issuance costs | 282 | — | |||||
Stock-based compensation expense |
| 3,379 |
| 5,035 | |||
Amortization of net contract asset/liability | (375) | — | |||||
Noncash lease expense |
| 80 |
| 73 | |||
Noncash lease liability | 69 | 82 | |||||
Write-off of fixed assets | 61 | — | |||||
Issuance of common stock for cost of license agreement | 1,168 | — | |||||
Unrealized loss on foreign currency transactions | 930 | — | |||||
Changes in operating assets and liabilities: | |||||||
Refund liability | (22,163) | — | |||||
Net contract asset/liability |
| 10,045 |
| — | |||
Prepaid expenses and other current assets, non-current assets, and accounts receivable |
| (1,072) |
| 366 | |||
Accounts payable, accrued expenses and other long term-liabilities |
| (783) |
| 5,325 | |||
Net cash used in operating activities |
| (27,655) |
| (16,175) | |||
Cash flows from investing activities | |||||||
Maturities of short-term investments |
| — |
| 1,474 | |||
Deposit on property and equipment | — | (404) | |||||
Purchases of property and equipment |
| (86) |
| (28) | |||
Net cash (used in) provided by investing activities |
| (86) |
| 1,042 | |||
Cash flows from financing activities | |||||||
Proceeds from exercise of stock options |
| 1,733 |
| 244 | |||
Proceeds from notes payable, net of upfront fee | 29,400 | — | |||||
Financing costs, paid | — | (148) | |||||
Net cash provided by financing activities |
| 31,133 |
| 96 | |||
Net increase (decrease) in cash and cash equivalents |
| 3,392 |
| (15,037) | |||
Cash and cash equivalents—beginning of period |
| 122,927 |
| 138,509 | |||
Cash and cash equivalents—end of period | $ | 126,319 | $ | 123,472 | |||
Supplemental disclosure of cash flow information | |||||||
Debt issuance costs included in accrued expenses | $ | 563 | $ | — | |||
Property and equipment in accrued expenses | $ | 205 | $ | — | |||
Property and equipment in deposits placed in service | $ | 337 | $ | — | |||
See accompanying notes to consolidated financial statements.
5
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(in thousands, except share amounts)
(unaudited)
Series A | Additional | Total | |||||||||||||||||||||||
Convertible Preferred Stock | Common Stock | Paid-in | Treasury Stock | Accumulated | Stockholders’ | ||||||||||||||||||||
| Shares |
| Amount |
| Shares |
| Amount |
| Capital |
| Shares |
| Amount |
| Deficit |
| Equity | ||||||||
Balance, December 31, 2020 | 4,753 | $ | 4,469 | 36,578,460 | $ | 37 | $ | 444,622 | 7,307 | $ | — | $ | (311,929) | $ | 137,199 | ||||||||||
Stock-based compensation expense | — | — | — | — | 5,035 | — | — | — | 5,035 | ||||||||||||||||
Exercise of stock options | — | — | 55,030 | — | 244 | — | — | — | 244 | ||||||||||||||||
Financing costs | — | — | — | — | (3) | — | — | — | (3) | ||||||||||||||||
Net Loss | — | — | — | — | — | — | — | (27,141) | (27,141) | ||||||||||||||||
Balance, March 31, 2021 |
| 4,753 | $ | 4,469 | 36,633,490 | $ | 37 | $ | 449,898 | 7,307 | $ | — | $ | (339,070) | $ | 115,334 | |||||||||
Balance, December 31, 2021 |
| 4,575 | $ | 4,302 | 36,790,254 | $ | 37 | $ | 459,852 | 7,307 | $ | — | $ | (410,705) | $ | 53,486 | |||||||||
Stock-based compensation expense | — | — | — | — | 3,379 | — | — | — | 3,379 | ||||||||||||||||
Exercise of stock options | — | — | 225,165 | — | 1,733 | — | — | — | 1,733 | ||||||||||||||||
Issuance of stock related to IP license agreement with Ovid | — | — | 123,255 | — | 1,168 | — | — | — | 1,168 | ||||||||||||||||
Net Loss | — | — | — | — | — | — | — | (19,361) | (19,361) | ||||||||||||||||
Balance, March 31, 2022 | 4,575 | $ | 4,302 | 37,138,674 | $ | 37 | $ | 466,132 | 7,307 | $ | — | $ | (430,066) | $ | 40,405 | ||||||||||
See accompanying notes to consolidated financial statements.
6
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED NOTES TO FINANCIAL STATEMENTS
1. Description of the Business and Liquidity
We are a pharmaceutical company focused on the development and commercialization of products for patients suffering from rare genetic epilepsies and other seizure disorders. On March 18, 2022, the U.S Food and Drug Administration (FDA) approved our new drug application (NDA) for the use of ZTALMY® (ganaxolone) oral suspension for the treatment of seizures associated with cyclin-dependent kinase-like 5 (CDKL5) deficiency disorder (CDD) in patients 2 years of age and older. This is our first FDA-approved product which we plan to commercialize in the United States. We also plan to continue to develop ganaxolone for other rare genetic epilepsies and for a number of additional indications including other seizure disorders.
The continued global spread of COVID-19, including the Omicron variant and its subvariants, has impacted our clinical operations and timelines. For example, our Phase 3 Randomized Therapy In Status Epilepticus Trial (RAISE trial) in refractory status epilepticus (RSE) is conducted in hospitals, including intensive care units and academic medical centers, which have experienced high rates of COVID-19 admissions. Several academic medical centers and intensive care units participating in the RAISE trial have experienced COVID-related difficulties, including staff turnover and the need to devote significant resources to patients with COVID-19, which has resulted in site initiation and enrollment delays for the RAISE trial. Given these COVID-19-related challenges and a temporary pause beginning in February 2022 of the RAISE trial after routine monitoring of stability batches of clinical supply material indicated that it became necessary to reduce the shelf life to less than the anticipated 24-months to meet product stability testing specifications, we now expect our top-line data readout for the RAISE trial to be available in the second half of 2023. In May 2022, we resumed screening and recruitment for the RAISE trial. In addition, our ganaxolone clinical trials in the outpatient setting may be negatively impacted if patients and their caregivers do not want to participate while the COVID-19 pandemic persists. The duration and severity of the pandemic and its long-term impact on our business are uncertain at this time.
Liquidity
We have not generated any product revenues and have incurred operating losses since inception, including losses of $19.4 million for the three months ended March 31, 2022. There is no assurance that profitable operations will ever be achieved, and if achieved, could be sustained on a continuing basis. In addition, development activities, clinical and preclinical testing, and commercialization of ganaxolone will require significant additional financing. Our accumulated deficit as of March 31, 2022 was $430.1 million and we expect to incur substantial losses in future periods. We plan to finance our future operations with a combination of proceeds from the issuance of equity securities, the issuance of debt, government funding, collaborations, licensing transactions and other commercial transactions or other sources, such as monetization of our Priority Review Voucher, and revenues from future product sales, if any. We have not generated positive cash flows from operations, and there are no assurances that we will be successful in obtaining an adequate level of financing for the commercialization and continued development of ganaxolone.
On July 30, 2021, we entered into a collaboration agreement (the Orion Collaboration Agreement) with Orion Corporation (Orion), whereby Orion received exclusive rights to commercialize the oral and IV dose formulations of ganaxolone in the European Economic Area, United Kingdom and Switzerland in multiple seizure disorders, including CDD, tuberous sclerosis complex (TSC) and RSE. Under the agreement, we received a €25 million ($29.6 million) upfront fee and are eligible to receive up to an additional €97 million in research and development reimbursement and cash milestone payments based on specific clinical and commercial achievements, as well as tiered royalty payments based on net sales ranging from the low double digits to the high teens for the oral programs and the low double-digits to the low twenties for the IV programs. In connection with the upfront fee, we agreed to provide Orion with the results of an ongoing genotoxicity study on the M2 metabolite of ganaxolone, a “Combined Micronucleus & Comet study in vivo.” In the event that the results of such study were positive, based on the criteria set forth in the study’s protocol, Orion would have had the right to terminate the Orion Collaboration Agreement within ninety () days after its receipt of the final report of such study, in which case we would have been required to refund Orion seventy-five percent (75%)
7
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED NOTES TO FINANCIAL STATEMENTS (Continued)
of the upfront fee. In the event of such termination and refund, Orion would have had no further rights pursuant to the oral and IV dose formulations of ganaxolone and the Orion Collaboration Agreement would have terminated and been of no further force or effect. In February 2022, the verified draft study report showed that no genotoxicity was found, as measured by formation of micronuclei in the bone marrow or comet morphology in the liver. These results were formalized in the final study report received in May 2022 and, as a result of the study’s findings, we are not required to refund Orion any of the upfront fee and Orion does not have the right to terminate the Orion Collaboration Agreement based on the study outcome.
On May 11, 2021 (Closing Date), we entered into a Credit Agreement and Guaranty (as amended by that certain letter agreement on May 17, 2021, the Credit Agreement) with Oaktree Fund Administration, LLC, as administrative agent and the lenders party thereto that provides for a senior secured term loan facility in an aggregate original principal amount of up to $125.0 million available to us in five tranches (collectively, the Term Loans). As of March 31, 2022, we have drawn a total of $75.0 million of the Term Loans pursuant to the Credit Agreement. Refer to Note 8. Notes Payable for additional information.
In September 2020, we entered into a contract (BARDA Contract) with the Biomedical Advanced Research and Development Authority (BARDA), a division of the U.S. Department of Health and Human Services’ Office of the Assistant Secretary for Preparedness and Response. Under the BARDA Contract, we received an award of up to an estimated $51 million for development of IV-administered ganaxolone for the treatment of RSE. The BARDA Contract provides for funding to support, on a cost-sharing basis, the completion of a Phase 3 clinical trial of IV-administered ganaxolone in patients with RSE, which covers the RAISE trial, funding of pre-clinical studies to evaluate IV-administered ganaxolone as an effective treatment for RSE due to chemical nerve gas agent exposure, and funding of certain ganaxolone manufacturing scale-up and regulatory activities. In March 2022, we entered into an amendment with BARDA to extend the end date of our base performance period for funding under the BARDA Contract from September 1, 2022 to December 31, 2023.
The BARDA Contract consists of an approximately base period, and an extension period through December 31, 2023 per the amendment described above, during which BARDA will provide up to approximately $21 million of funding for the RAISE trial on a cost share basis and funding of additional preclinical studies of ganaxolone in nerve agent exposure models. Following successful completion of the RAISE trial and preclinical studies in the base period and extension period, the BARDA Contract provides for approximately $30 million of additional BARDA funding for three options in support of ganaxolone manufacturing, supply chain, clinical, regulatory and toxicology activities. Under the BARDA Contract, we will be responsible for cost sharing in the amount of approximately $33 million and BARDA will be responsible for approximately $51 million, if all development options are completed. The contract period-of-performance (base and extension periods plus option exercises) is up to approximately .
Management’s operating plan, which underlies the analysis of our ability to continue as a going concern, involves the estimation of the amount and timing of future cash inflows and outflows. Actual results could vary from the operating plan. We follow the provisions of Financial Accounting Standards Board (FASB) Accounting Standards Codification (ASC) Topic 205-40, Presentation of Financial Statements—Going Concern, which requires management to assess our ability to continue as a going concern within one year after the date the financial statements are issued. Our cash and cash equivalents on hand as of March 31, 2022, excluding the $15.0 million liquidity requirement associated with our Note Payable (Note 8), is not sufficient to fund operations for the one-year period after the date the financial statements are issued. As a result, there is substantial doubt about our ability to continue as a going concern through the one-year period from the date these financial statements are issued. Management’s plans that are intended to mitigate this risk include the monetization of our Priority Review Voucher and additional financing or strategic transactions. We have and will continue to evaluate available alternatives to further extend our operations beyond the one-year period after the date the financial statements are issued. If additional capital is not obtained when required, we may need to delay or curtail our operations until additional funding is received.
8
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED NOTES TO FINANCIAL STATEMENTS (Continued)
2. Summary of Significant Accounting Policies
Basis of Presentation
The accompanying unaudited interim consolidated financial statements include the accounts of Marinus Pharmaceuticals, Inc. (a Delaware corporation) as well as the accounts of Marinus Pharmaceuticals Emerald Limited (an Ireland company incorporated in February 2021), a wholly owned subsidiary requiring consolidation. Marinus Pharmaceuticals Emerald Limited serves as a corporate presence in the European Union for regulatory purposes. The unaudited interim consolidated financial statements included herein have been prepared pursuant to the rules and regulations of the Securities and Exchange Commission (SEC). Accordingly, they do not include all information and disclosures necessary for a presentation of our financial position, results of operations and cash flows in conformity with generally accepted accounting principles in the U.S. (GAAP) for annual financial statements. In the opinion of management, these unaudited interim consolidated financial statements reflect all adjustments, consisting primarily of normal recurring accruals, necessary for a fair presentation of our financial position and results of operations and cash flows for the periods presented. The results of operations for interim periods are not necessarily indicative of the results for the full year. These unaudited interim consolidated financial statements should be read in conjunction with the audited financial statements for the year ended December 31, 2021 and accompanying notes thereto included in our Annual Report on Form 10-K filed with the SEC on March 24, 2022.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of income and expenses during the reporting period. Actual results could differ from such estimates.
Federal Contract Revenue
We recognize federal contract revenue from the BARDA Contract in the period in which the allowable research and development expenses are incurred, and receivables associated with this revenue are included within accounts receivable on our interim consolidated balance sheets. This revenue is not within the scope of ASC 606 – Revenue from contracts with customers.
Debt Issuance Costs
Debt issuance costs incurred in connection with Note payable (Note 8) are amortized to interest expense over the term of the respective financing arrangement using the effective-interest method. Debt issuance costs, net of related amortization are deducted from the carrying value of the related debt.
Contract Liability
When consideration is received, or such consideration is unconditionally due, from a customer prior to completing our performance obligation to the customer under the terms of a contract, a contract liability is recorded. Contract liabilities expected to be recognized as revenue or a reduction of expense within the 12 months following the balance sheet date are classified as current liabilities. Contract liabilities not expected to be recognized as revenue within the 12 months following the balance sheet date are classified as long-term liabilities. In accordance with ASC 210-20, our contract liability is partially offset by a contract asset as further discussed in Note 9.
9
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED NOTES TO FINANCIAL STATEMENTS (Continued)
Collaboration and Licensing Revenue
We may enter into collaboration and licensing arrangements for research and development, manufacturing, and commercialization activities with counterparties for the development and commercialization of our product candidates. These arrangements may contain multiple components, such as (i) licenses, (ii) research and development activities, and (iii) the manufacturing of certain material. Payments pursuant to these arrangements may include non-refundable and refundable payments, payments upon the achievement of significant regulatory, development and commercial milestones, sales of product at certain agreed-upon amounts, and royalties on product sales. The amount of variable consideration is constrained until it is probable that the revenue is not at a significant risk of reversal in a future period.
In determining the appropriate amount of revenue to be recognized as we fulfill our obligations under a collaboration agreement, we perform the following steps: (i) identification of the promised goods or services in the contract; (ii) determination of whether the promised goods or services are performance obligations, including whether they are capable of being distinct; (iii) measurement of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition of revenue as we satisfy each performance obligation.
We must develop estimates and assumptions that require judgment to determine the underlying stand-alone selling price for each performance obligation, which determines how the transaction price is allocated among the performance obligations. The estimation of the stand-alone selling price may include such estimates as forecasted revenues and costs, development timelines, discount rates and probabilities of regulatory and commercial success. We also apply significant judgment when evaluating whether contractual obligations represent distinct performance obligations, allocating transaction price to performance obligations within a contract, determining when performance obligations have been met, assessing the recognition and future reversal of variable consideration and determining and applying appropriate methods of measuring progress for performance obligations satisfied over time.
3. Fair Value Measurements
FASB accounting guidance defines fair value as the price that would be received to sell an asset or paid to transfer a liability (the exit price) in an orderly transaction between market participants at the measurement date. The accounting guidance outlines a valuation framework and creates a fair value hierarchy in order to increase the consistency and comparability of fair value measurements and the related disclosures. In determining fair value, we use quoted prices and observable inputs. Observable inputs are inputs that market participants would use in pricing the asset or liability based on market data obtained from independent sources.
The fair value hierarchy is broken down into three levels based on the source of inputs as follows:
| ● | Level 1 — Valuations based on unadjusted quoted prices in active markets for identical assets or liabilities. |
| ● | Level 2 — Valuations based on observable inputs and quoted prices in active markets for similar assets and liabilities. |
| ● | Level 3 — Valuations based on inputs that are unobservable and models that are significant to the overall fair value measurement. |
If the inputs used to measure fair value fall within different levels of the hierarchy, the category level is based on the lowest priority level input that is significant to the fair value measurement of the instrument. As of March 31, 2022 and December 31, 2021, all of our financial assets and liabilities were classified as Level 1 valuations.
10
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED NOTES TO FINANCIAL STATEMENTS (Continued)
The following fair value hierarchy table presents information about each major category of our financial assets and liabilities measured at fair value on a recurring basis (in thousands):
| Level 1 |
| Level 2 |
| Level 3 |
| Total |
| |||||
March 31, 2022 | |||||||||||||
Assets | |||||||||||||
Cash | $ | 34,128 | $ | — | $ | — | $ | 34,128 | |||||
Money market funds (cash equivalents) | 92,191 | — | — | 92,191 | |||||||||
Total assets | $ | 126,319 | $ | — | $ | — | $ | 126,319 | |||||
December 31, 2021 | |||||||||||||
Assets | |||||||||||||
Cash | $ | 2,360 | $ | — | $ | — | $ | 2,360 | |||||
Money market funds (cash equivalents) | 120,567 | — | — | 120,567 | |||||||||
Total assets | $ | 122,927 | $ | — | $ | — | $ | 122,927 | |||||
4. Accrued Expenses
Accrued expenses consisted of the following (in thousands):
March 31, | December 31, | ||||||
2022 | 2021 | ||||||
Payroll and related costs | $ | 3,360 | $ | 5,830 |
| ||
Clinical trials and drug development | 8,765 | 8,217 | |||||
Professional fees | 1,431 | 1,311 | |||||
Debt issuance costs and commitment fees | 630 | — | |||||
Short-term lease liabilities | 575 | 556 | |||||
Other | 104 | 293 | |||||
Total accrued expenses | $ | 14,865 | $ | 16,207 | |||
5. Loss Per Share of Common Stock
Basic loss per share is computed by dividing net loss applicable to common stockholders by the weighted average number of shares of common stock outstanding during each period. Diluted loss per share includes the effect, if any, from the potential exercise or conversion of securities, such as convertible preferred stock, convertible notes payable, warrants, stock options, and unvested restricted stock, which would result in the issuance of incremental shares of common stock. In computing the basic and diluted net loss per share applicable to common stockholders, the weighted average number of shares remains the same for both calculations due to the fact that when a net loss exists, dilutive shares are not included in the calculation. These potentially dilutive securities are more fully described in Note 6, and summarized in the table below:
March 31, | |||||
2022 | 2021 | ||||
Convertible preferred stock |
| 915,000 |
| 950,600 |
|
Restricted stock awards and restricted stock units |
| 541,670 |
| 22,000 |
|
Stock options |
| 5,654,024 |
| 4,436,778 |
|
| 7,110,694 |
| 5,409,378 |
| |
11
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED NOTES TO FINANCIAL STATEMENTS (Continued)
6. Stockholders’ Equity
In 2005, we adopted the 2005 Stock Option and Incentive Plan (2005 Plan) that authorizes us to grant stock options, restricted stock and other equity-based awards. As of March 31, 2022, 577 options to purchase shares of common stock were outstanding pursuant to grants in connection with the 2005 Plan. No additional shares are available for issuance under the 2005 Plan. The amount, terms of grants, and exercisability provisions are determined and set by our board of directors.
Effective August 2014, we adopted our 2014 Equity Incentive Plan, as amended (2014 Plan), that authorizes us to grant stock options, restricted stock, and other equity-based awards, subject to adjustment in accordance with the 2014 Plan. As of March 31, 2022, 4,072,818 options to purchase shares of common stock were outstanding pursuant to grants in connection with the 2014 Plan, and 816,377 shares of common stock were available for future issuance. The amount, terms of grants, and exercisability provisions are determined and set by our board of directors. In accordance with the 2014 Plan, on January 1, 2022, the shares of common stock available for future grants under the 2014 Plan was increased to 2,343,330.
Stock Options
There were 5,654,024 stock options outstanding as of March 31, 2022 at a weighted average exercise price of $11.66 per share, including 1,580,629 stock options outstanding outside of the 2014 Plan, granted as inducements to new employees. During the three months ended March 31, 2022, 1,341,399 options were granted to employees and directors at a weighted average exercise price of $10.47 per share. Of the options granted, 1,204,388 options were granted pursuant to the 2014 Plan and 137,011 were granted outside of the 2014 Plan as inducements for new employees.
Total compensation cost recognized for all stock option awards in the statements of operations is as follows (in thousands):
Three Months Ended | ||||||
March 31, | ||||||
2022 | 2021 | |||||
Research and development |
| $ | 1,179 |
| $ | 1,190 |
General and administrative |
| 1,899 |
| 3,790 | ||
Total | $ | 3,078 | $ | 4,980 | ||
Restricted Stock
All issued and outstanding restricted shares of common stock are time-based, and become vested within two years of the grant date, pursuant to the 2014 Plan. Compensation expense is recorded ratably over the requisite service period. Compensation expense related to restricted stock is measured based on the fair value using the closing market price of our common stock on the date of the grant. As of March 31, 2022, we had 22,625 outstanding shares of restricted common stock.
During the three months ended March 31, 2022, we granted 515,645 restricted stock units, which vest within four years of the grant date, pursuant to the 2014 Plan. As of March 31, 2022, we had 519,045 restricted stock units outstanding.
12
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED NOTES TO FINANCIAL STATEMENTS (Continued)
Total compensation cost recognized for all restricted stock awards and restricted stock units in the statements of operations is as follows (in thousands):
Three Months Ended |
| |||||||
March 31, |
| |||||||
2022 | 2021 |
| ||||||
Research and development |
| $ | 99 |
| $ | — |
| |
General and administrative |
| 202 |
| 55 | ||||
Total | $ | 301 | $ | 55 | ||||
Preferred Stock
As of March 31, 2022, 4,575 shares of our Series A Convertible Preferred Stock (Preferred Stock) remained outstanding, convertible into 915,000 shares of our common stock.
Stock Issued in Connection with Ovid License Agreement
On March 29, 2022, pursuant to an exclusive patent license agreement with Ovid Therapeutics Inc. (Ovid), we issued 123,255 shares of our common stock to Ovid. The shares were issued in reliance upon the exemption from the registration requirements of the Securities Act of 1933, as amended (the Securities Act) provided by Section 4(a)(2) of the Securities Act and Regulation D thereunder as sales by an issuer not involving any public offering (see Part II, Item 2. Unregistered Sales of Equity Securities and Use of Proceeds). The fair value of these shares is reflected in operating expenses for the three months ended March 31, 2022.
7. Leases
We have entered into one operating lease for real estate. This lease has a term of 78 months, and includes renewal terms which can extend the lease terms by 60 months (which we include in lease terms when it is reasonably certain that we will exercise the option). As of March 31, 2022, our operating lease had a remaining lease term of 42 months. The right-of-use (ROU) asset is included in "Other assets" on our interim consolidated balance sheets as of March 31, 2022 and December 31, 2021, and represents our right to use the underlying asset for the lease term. Our obligations to make lease payments are included in "" and "" on our interim consolidated balance sheets as of March 31, 2022 and December 31, 2021, respectively. The ROU asset was initially measured at cost, which comprises the initial amount of the lease liability adjusted for lease payments made at or before the lease commencement date, plus any initial direct costs incurred, less any lease incentives received. The ROU asset is subsequently measured throughout the lease term at the carrying amount of the lease liability, plus initial direct costs, plus (minus) any prepaid (accrued) lease payments, less the unamortized balance of lease incentives received.
As of March 31, 2022 and December 31, 2021, ROU assets were $1.6 million and $1.7 million, respectively, and operating lease liabilities were $2.4 million and $2.5 million, respectively. We have entered into various short-term operating leases, primarily for clinical trial equipment, with an initial term of twelve months or less. These leases are not recorded on our balance sheets. All operating lease expense is recognized on a straight-line basis over the lease term. During the three months ended March 31, 2022 and 2021, we recognized $0.1 million and $0.2 million, respectively, in total lease costs, each of which included less than $0.1 million in short-term lease costs related to short-term operating leases.
Because the rate implicit in each lease is not readily determinable, we use our incremental borrowing rate to determine the present value of the lease payments. The weighted average incremental borrowing rate used to determine the initial value of ROU assets and lease liabilities was 11.0%, derived from a corporate yield curve based on a synthetic
13
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED NOTES TO FINANCIAL STATEMENTS (Continued)
credit rating model using a market signal analysis. We have certain contracts for real estate which may contain lease and non-lease components which we have elected to treat as a single lease component.
ROU assets for operating leases are periodically reduced by impairment losses. We use the long-lived assets impairment guidance in ASC Subtopic 360-10, Property, Plant, and Equipment – Overall, to determine whether an ROU asset is impaired, and if so, the amount of the impairment loss to recognize. As of March 31, 2022 and December 31, 2021, we have not recognized any impairment losses for our ROU assets.
We monitor for events or changes in circumstances that require a reassessment of our lease. When a reassessment results in the remeasurement of a lease liability, a corresponding adjustment is made to the carrying amount of the corresponding ROU asset unless doing so would reduce the carrying amount of the ROU asset to an amount less than zero. In that case, the amount of the adjustment that would result in a negative ROU asset balance is recorded in our interim consolidated statements of operations and comprehensive loss.
Maturities of operating lease liabilities as of March 31, 2022 were as follows (in thousands):
|
| |||
Remaining nine months of 2022 | $ | 609 | ||
2023 |
| 823 | ||
2024 |
| 840 | ||
2025 |
| 642 | ||
Thereafter |
| — | ||
2,914 | ||||
Less: imputed interest | (510) | |||
Total lease liabilities | $ | 2,404 | ||
Current operating lease liabilities | $ | 575 | ||
Non-current operating lease liabilities | 1,829 | |||
Total lease liabilities | $ | 2,404 | ||
8. Notes Payable
On May 11, 2021 (Closing Date) and as amended on May 17, 2021, we entered into the Credit Agreement with Oaktree Fund Administration, LLC as administrative agent (Oaktree) and the lenders party thereto (collectively, the Lenders) that provides for a five-year senior secured term loan facility in an aggregate original principal amount of up to $125.0 million, available to us in five tranches (collectively, the Term Loans).
Upon entering into the Credit Agreement in May 2021, we borrowed $15.0 million in term loans from the Lenders (Tranche A-1 Term Loan); upon receipt of written acceptance by the FDA of our NDA filing relating to the use of ganaxolone in CDD in September 2021 we borrowed $30.0 million of tranche A-2 term loans from the Lenders (Tranche A-2 Term Loan); and in March 2022, we borrowed $30.0 million in term loans from the Lenders that became available as a result of the approval by the FDA of ZTALMY oral suspension for the treatment of seizures associated with CDD in patients two years of age and older (Tranche B Term Loan). Under the terms of the Credit Agreement, we may, at our sole discretion, borrow from the Lenders up to an additional $50.0 million in term loans subject to certain milestone events, as follows:
| ● | Through June 30, 2023, $25.0 million of tranche C term loans will be available for draw if we complete one or more financings (including through the issuance of common stock, convertible debt, subordinated debt, a synthetic royalty, or a sublicense) resulting in gross proceeds to us of at least $40.0 million and net |
14
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED NOTES TO FINANCIAL STATEMENTS (Continued)
| proceeds to us of at least $36.0 million. In addition, the availability of this tranche is subject to certain clinical outcomes. |
| ● | Through December 31, 2023, $25.0 million of tranche D term loans will be available for draw if we earn an aggregate of at least $50.0 million in net product revenue in the U.S. for a trailing consecutive months. |
In addition, the Credit Agreement contains a minimum liquidity covenant that requires us to maintain cash and cash equivalents of at least $15.0 million from the funding date of the tranche B term loans until the maturity of the Term Loans.
The Term Loans will be guaranteed by certain of our future subsidiaries (Guarantors). Our obligations under the Credit Agreement are secured by a pledge of substantially all of our assets and will be secured by a pledge of substantially all of the assets of the Guarantors.
The Term Loans mature on May 11, 2026 (Maturity Date). The Term Loans bear interest at a fixed per annum rate (subject to increase during an event of default) of 11.50%, and we are required to make quarterly interest payments until the Maturity Date. We are also required to make quarterly principal payments beginning on June 30, 2024 in an amount equal to 5.0% of the aggregate amount of the Term Loans outstanding on June 30, 2024, and continuing until the Maturity Date. On the Maturity Date, we are required to pay in full all outstanding Term Loans and other amounts owed under the Credit Agreement.
At the time of borrowing any tranche of the Term Loans, we are required to pay an upfront fee of 2.0% of the aggregate principal amount borrowed at that time. In addition, a commitment fee of 75 basis points per annum began to accrue on each of the tranche B, C, and D commitments for the period beginning after the funding date of the tranche A-2 term loans, and continues until the applicable tranche is either funded or terminated, at which time the related commitment fees are due. The tranche A-2 term loans were funded on September 27, 2021, and as such, we began accruing the commitment fees for tranche B, C, and D term loans 120 days later, on January 25, 2022. We drew down the additional $30.0 million of tranche B term loans in March 2022, and paid less than $0.1 million in commitment fees related to tranche B term loans. At March 31, 2022, we had less than $0.1 million accrued for commitment fees related to tranche C and D term loans.
We may prepay all or any portion of the Term Loans, and are required to make mandatory prepayments of the Term Loans from the proceeds of asset sales, casualty and condemnation events, and prohibited debt issuances, subject to certain exceptions. All mandatory and voluntary prepayments of the Term Loans are subject to prepayment premiums equal to (i) 4% of the principal prepaid plus a “make-whole” amount equal to the interest that would have accrued through May 11, 2023 if prepayment occurs on or before May 11, 2023, (ii) 4% of the principal prepaid if prepayment occurs after May 11, 2023 but on or before May 11, 2024, or (iii) 2% of the principal prepaid if prepayment occurs after May 11, 2024 but on or before May 11, 2025. If prepayment occurs after May 11, 2025, no prepayment premium is due.
We are also required to make mandatory prepayments of the Term Loans upon an event of default under the Credit Agreement resulting from the occurrence of a change of control. These mandatory prepayments are subject to a prepayment premium equal to (i) 12.5% of the principal prepaid if such prepayment occurs on or before May 11, 2022 or (ii) 10.0% of the principal prepaid if the prepayment occurs after May 11, 2022 but on or before May 11, 2023.
In addition, we are required to pay an exit fee in an amount equal to 2.0% of all principal repaid, whether as a mandatory prepayment, voluntary prepayment, or a scheduled repayment.
In addition to the minimum liquidity covenant, we are subject to a number of affirmative and restrictive covenants under the Credit Agreement, including limitations on our ability and our subsidiaries’ abilities, among other things, to incur additional debt, grant or permit additional liens, make investments and acquisitions, merge or consolidate
15
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED NOTES TO FINANCIAL STATEMENTS (Continued)
with others, dispose of assets, pay dividends and distributions, and enter into affiliate transactions, subject to certain exceptions. As of March 31, 2022, we were in compliance with all covenants.
Upon the occurrence of certain events, including but not limited to our failure to satisfy our payment obligations under the Credit Agreement, the breach of certain of our other covenants under the Credit Agreement, the occurrence of cross defaults to other indebtedness, or defaults related to enforcement action by the FDA or other Regulatory Authority or recall of ganaxolone, Oaktree and the Lenders will have the right, among other remedies, to accelerate all amounts outstanding under the Term Loans and declare all principal, interest, and outstanding fees immediately due and payable.
In March 2022, we borrowed $30.0 million upon the approval by the FDA of ZTALMY for CDD and incurred debt issuance costs of $1.8 million, including the exit fee of $0.6 million, that are classified as contra-liabilities on our consolidated balance sheets and are being recognized as interest expenses over the term of the loan using the effective interest method.
In September 2021, we borrowed $30.0 million upon receipt of written acceptance by the FDA of our NDA filing relating to the use of ganaxolone in the treatment of CDD and incurred debt issuance costs of $1.2 million, including the exit fee of $0.6 million, that are classified as contra-liabilities on our consolidated balance sheets and are being recognized as interest expenses over the term of the loan using the effective-interest method.
In May 2021, we borrowed $15.0 million upon entering into the Credit Agreement and incurred debt issuance costs of $4.4 million, including the exit fee of $0.3 million, that are classified as a contra-liabilities on the consolidated balance sheet and are being recognized as interest expenses over the term of the loan using the effective-interest method.
For the three months ended March 31, 2022, we recognized interest expense of $1.7 million, of which $1.3 million was interest on the Term Loans, $0.3 million was non-cash interest expense related to the amortization of debt issuance costs, less than $0.1 million was non-cash interest expense related to the commitment fee, and less than $0.1 million was related to commitment fees paid in the three months ended March 31, 2022.
The following table summarizes the composition of Notes payable as reflected on the consolidated balance sheet as of March 31, 2022 (in thousands):
Gross proceeds | $ | 75,000 | |
Contractual exit fee |
| 1,500 | |
Unamortized debt discount and issuance costs |
| (6,573) | |
Total | $ | 69,927 |
The aggregate maturities of Notes payable as of March 31, 2022 are as follows (in thousands):
Remainder of 2022 | $ | — | |
2023 | — | ||
2024 | 11,250 | ||
2025 | 15,000 | ||
2026 and thereafter | 48,750 | ||
Total | $ | 75,000 |
16
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED NOTES TO FINANCIAL STATEMENTS (Continued)
9. Collaboration Revenue
In July 2021, we entered into a collaboration agreement (the Orion Collaboration Agreement) with Orion Corporation (Orion). The Orion Collaboration Agreement falls under the scope of ASC Topic 808, Collaborative Arrangements (ASC 808) as both parties are active participants in the arrangement that are exposed to significant risks and rewards. While this arrangement is in the scope of ASC 808, we analogize to ASC 606 for some aspects of this arrangement, including for the delivery of a good or service (i.e., a unit of account). Revenue recognized by analogizing to ASC 606 is recorded as collaboration revenue on the consolidated statements of operations.
Under the terms of the Orion Collaboration Agreement, we granted Orion an exclusive, royalty-bearing, sublicensable license to certain of our intellectual property rights with respect to commercializing biopharmaceutical products incorporating our product candidate ganaxolone (Licensed Products) in the European Economic Area, the United Kingdom and Switzerland (collectively, the Territory) for the diagnosis, prevention and treatment of certain human diseases, disorders or conditions (Field), initially in the indications of CDD, TSC and RSE. We will be responsible for the continued development of Licensed Products and regulatory interactions related thereto, including conducting and sponsoring all clinical trials, provided that Orion may conduct certain post-approval studies in the Territory. Orion will be responsible, at Orion’s sole cost and expense, for the commercialization of any Licensed Product in the Field in the Territory.
Under the terms of the Orion Collaboration Agreement, we received a €25.0 million ($29.6 million) upfront payment from Orion in July 2021. In connection with the upfront fee, we agreed to provide Orion with the results of a planned genotoxicity study on the M2 metabolite of ganaxolone, a “Combined Micronucleus & Comet study in vivo.” In the event that the results of such study were positive, based on the criteria set forth in the study’s protocol, Orion would have had the right to terminate the Orion Collaboration Agreement within ninety () days after its receipt of the final report of such study, in which we would have been required to refund Orion seventy-five percent (75%) of the upfront fee. In the event of such termination and refund, Orion would have had no further rights pursuant to the oral and IV dose formulations of ganaxolone and the Orion Collaboration Agreement would have terminated and be of no further force or effect. In February 2022, the verified draft study report showed that no genotoxicity was found, as measured by formation of micronuclei in the bone marrow or comet morphology in the liver. These results were formalized in the final study report received in May 2022 and, as a result of the study’s findings, we are not required to refund Orion any of the upfront fee and Orion does not have the right to terminate the Orion Collaboration Agreement based upon the study outcome. We are eligible to receive up to an additional €97 million in research and development reimbursement and cash milestone payments based on specific clinical and commercial achievements, as well as tiered royalty payments based on net sales ranging from the low double-digits to high teens for the oral programs and the low double-digits to low 20s for the IV program. Also, as part of the overall arrangement, we have agreed to supply the Licensed Products to Orion at an agreed upon price.
The Orion Collaboration Agreement shall remain effective until the date of expiration of the last to expire Royalty Term, which is defined as the period beginning on the date of the first commercial sale Licensed Product in such country and ending on the latest to occur of (a) the tenth (10th) anniversary of the first commercial sale of Licensed Product in such country, (b) the expiration of the last-to-expire licensed patent covering the manufacture, use or sale of such Licensed Product in such country, and (c) the expiration of regulatory exclusivity period, if any, for such Licensed Product in such country. The Orion Collaboration Agreement has a term of at least ten () years since a commercial sale has yet to occur. The Orion Collaboration Agreement allows for termination in certain specific events, such as material breach, in the event Orion challenges the validity, enforceability or scope of the licensed patent rights, termination for forecast failure, insolvency and force majeure, none of which are probable at contract inception.
In accordance with the guidance, we identified the following commitments under the arrangement: (i) exclusive rights to develop, use, sell, have sold, offer for sale and import any product comprised of Licensed Product (License), (ii) development and regulatory activities (Development and Regulatory Activities), and (iii) requirement to supply Orion
17
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED NOTES TO FINANCIAL STATEMENTS (Continued)
with the Licensed Product at an agreed upon price (Supply of Licensed Product). We determined that these three commitments represent distinct performance obligations for purposes of recognizing revenue or reducing expense, which we will recognize such revenue or expense, as applicable, as we fulfill these performance obligations.
At contract inception, we determined that the non-refundable portion of the upfront payment plus the research and development reimbursement constitutes the transaction price as of the outset of the Orion Collaboration Agreement. The refundable portion of the upfront payment and the future potential regulatory and development milestone payments were fully constrained at contract inception as the risk of significant revenue reversal related to these amounts had not yet been resolved. During the three months ended March 31, 2022, the refundable portion of the upfront payment was determined to be included in the transaction price as the final genotoxicity study on the M2 metabolite of ganaxolone was received as described above. The achievement of the future potential milestones is not within our control and is subject to certain research and development success and therefore carry significant uncertainty. We will reevaluate the likelihood of achieving these milestones at the end of each reporting period and adjust the transaction price in the period the risk is resolved. In addition, we will recognize any consideration related to sales-based milestones and royalties when the subsequent sales occur since those payments relate primarily to the License, which was delivered by us to Orion upon entering into the Orion Collaboration Agreement. We recorded $12.7 million of the $21.2 million refundable portion of the upfront payment as collaboration revenue in the three months ended March 31, 2022, and $9.5 million was recorded as a long-term liability as of March 31, 2022.
The transaction price was allocated to the three performance obligations based on the estimated stand-alone selling prices at contract inception. The stand-alone selling price of the License was based on a discounted cash flow approach and considered several factors including, but not limited to, discount rate, development timeline, regulatory risks, estimated market demand and future revenue potential using an adjusted market approach. The stand-alone selling price of the Development and Regulatory Activities and the Supply of Licensed Product was estimated using the expected cost-plus margin approach.
As of the agreement date in July 2021, we allocated the transaction price to the performance obligations as described below and recorded the $9.0 million transaction price associated with the License as revenue. During 2021, we amortized $0.1 million of the transaction price associated with the Development and Regulatory Services as a reduction of research and development costs. These reductions to transaction price resulted in a total contract liability of $6.6 million at December 31, 2021. In accordance with ASC 210-20, the contract liability of $6.6 million was offset by a contract asset of $7.2 million related to the reimbursement of research and development costs, resulting in a net contract asset of $0.6 million at December 31, 2021.
Transaction Price and Net Contract Asset at December 31, 2021:
Cumulative Collaboration | ||||||||
Transaction | Revenue Recognized | Contract | ||||||
Price |
| as of December 31, 2021 |
| Liability | ||||
License | $ | 8,987 | $ | 8,987 | $ | - | ||
Development and Regulatory Services | 2,787 | 106 | 2,681 | |||||
Supply of Licensed Product | 3,943 | - | 3,943 | |||||
$ | 15,717 | $ | 9,093 | 6,624 | ||||
Less Total Contract Asset | 7,181 | |||||||
Net Contract Asset | $ | 557 | ||||||
During the three months ended March 31, 2022, the refundable portion of the upfront payment was determined to be included in the transaction price as the final genotoxicity study on the M2 metabolite of ganaxolone was received as described above. As such, the refundable portion of the upfront payment of €18.8 million ($21.2 million) was allocated to the purchase price as shown below, resulting in a total purchase price of $37.9 million. Of the $21.2 million
18
MARINUS PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED NOTES TO FINANCIAL STATEMENTS (Continued)
refundable portion of the upfront payment, we recorded $12.7 million of collaboration revenue in the three months ended March 31, 2022. During the three months ended March 31, 2022, we amortized $0.4 million of the transaction price associated with the Development and Regulatory Services as a reduction of research and development costs. These reductions to the transaction price resulted in a total contract liability of $15.7 million at March 31, 2022. In accordance with ASC 210-20, the contract liability of $15.7 million is offset by a contract asset of $6.6 million related to the reimbursement of research and development costs, resulting in a net contract liability of $9.1 million at March 31, 2022.
Transaction Price and Net Contract Liability at March 31, 2022:
Cumulative Collaboration | ||||||||
Transaction Price | Revenue Recognized | Contract | ||||||
As of March 31, 2022 |
| as of March 31, 2022 |
| Liability | ||||
License | $ | 21,660 | $ | 21,660 | $ | - | ||
Development and Regulatory Services | 6,717 | 481 | 6,236 | |||||
Supply of Licensed Product | 9,503 | - | 9,503 | |||||
$ | 37,880 | $ | 22,141 | 15,739 | ||||
Less Total Contract Asset | 6,626 | |||||||
Net Contract Liability | $ | 9,113 | ||||||
In 2021, we incurred $2.0 million of incremental costs in obtaining the Orion Collaboration Agreement. These contract acquisition costs were allocated consistent with the transaction price, resulting in $1.1 million of expense recorded to general and administrative expense commensurate with the recognition of the License performance obligation and $0.9 million recorded as capitalized contract costs, included in other current assets and other assets, which are being amortized as Development and Regulatory Services and Supply of Licensed Product obligations are met.
We reevaluate the transaction price and the total estimated costs expected to be incurred to satisfy the performance obligations and adjusts the deferred revenue at the end of each reporting period. Such changes will result in a change to the amount of collaboration revenue recognized and deferred revenue.
19
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations
Cautionary Note Regarding Forward-Looking Statements
This Quarterly Report on Form 10-Q contains forward-looking statements, within the meaning of the U.S. Private Securities Litigation Reform Act of 1995, that involve substantial risks and uncertainties. In some cases, you can identify forward-looking statements by the words “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “might,” “objective,” “ongoing,” “plan,” “predict,” “project,” “potential,” “should,” “will,” or “would,” and or the negative of these terms, or other comparable terminology intended to identify statements about the future. These statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements. Although we believe that we have a reasonable basis for each forward-looking statement contained in this Quarterly Report on Form 10-Q, we caution you that these statements are based on a combination of facts and factors currently known by us and our expectations of the future, about which we cannot be certain.
The forward-looking statements in this Quarterly Report on Form 10-Q include, among other things, statements about:
| ● | our plans to successfully commercialize ganaxolone in Cyclin-dependent Kinase-like 5 Deficiency Disorder (CDD) in the United States; |
| ● | our plans to meet our post-approval commitments to the U.S. Food and Drug Administration (FDA) for ganaxolone; |
| ● | our plans to achieve regulatory approval for ganaxolone in the European Union (EU), and the expected timing thereof; |
| ● | our ability to develop ganaxolone for additional indications, including Refractory Status Epilepticus (RSE), Established Status Epilepticus (ESE), Tuberous Sclerosis Complex (TSC) and Lennox Gastaut Syndrome (LGS); |
| ● | the status, timing and results of preclinical studies and clinical trials; |
| ● | the design of and enrollment in clinical trials, availability of data from ongoing clinical trials, expectations for regulatory approvals, or the attainment of clinical trial results that will be supportive of regulatory approvals; |
| ● | the potential benefits of ganaxolone, including in indications other than CDD; |
| ● | the timing of seeking marketing approval of ganaxolone in specific additional indications; |
| ● | our ability to maintain marketing approval for ganaxolone in CDD and obtain regulatory approval for ganaxolone in other indications; |
| ● | the possibility that we expand the targeted indication footprint and explore new potential formulations of ganaxolone; |
| ● | our estimates of expenses and future revenue and profitability; |
| ● | our estimates regarding our capital requirements and our needs for additional financing; |
| ● | our estimates of the size of the potential markets for ganaxolone; |
20
| ● | our expectations regarding our collaboration with Orion Corporation (Orion), including the expected amount and timing of research and development reimbursement, milestone, royalty and other payments pursuant thereto; |
| ● | our ability to attract collaborators with acceptable development, regulatory and commercial expertise; |
| ● | the benefits to be derived from corporate collaborations, license agreements, and other collaborative or acquisition efforts, including those relating to the development and commercialization of ganaxolone; |
| ● | sources of revenue, including expected future sales of ganaxolone in CDD, revenue contributions from our contract (BARDA Contract) with the Biomedical Advanced Research and Development Authority (BARDA), corporate collaborations, license agreements, and other collaborative efforts for the development and commercialization of ganaxolone for CDD and in other indications being developed for ganaxolone; |
| ● | our eligibility to receive funding under the debt tranches available under the Credit Agreement with Oaktree; |
| ● | our ability to create an effective sales and marketing infrastructure where we elect to market and sell ganaxolone directly; |
| ● | the timing and amount of reimbursement for ganaxolone; |
| ● | the success of other competing therapies that may become available; |
| ● | the manufacturing capacity and supply for ganaxolone; |
| ● | the extent to which our ability to market and sell ganaxolone may be negatively impacted by third party patents; |
| ● | the possibility that we expand and diversify our product pipeline through acquisitions of additional drug candidates that fit our business strategy; |
| ● | our belief that our existing cash and cash equivalents will be sufficient to fund our operating expenses, capital expenditure requirements, and maintain the minimum cash balance required under our debt facility into the first quarter of 2023; |
| ● | our ability to maintain and protect our intellectual property rights; |
| ● | our expectations regarding the scheduling of ZTALMY by the U.S. Drug Enforcement Administration (DEA) as a controlled substance under the Controlled Substances Act of 1970 (CSA), and the timing thereof; |
| ● | our results of operations, financial condition, liquidity, prospects, and growth strategies; |
| ● | our ability to, among other actions, monetize our Priority Review Voucher, secure additional financing and/or secure strategic transactions and continue as a going concern; |
| ● | the extent to which our business may be adversely impacted by the effects of the COVID-19 coronavirus pandemic or by other pandemics, epidemics or outbreaks; |
21
| ● | the enforceability of the exclusive forum provisions in our fourth amended and restated certificate of incorporation; and |
| ● | the industry in which we operate and trends which may affect the industry or us. |
You should refer to Part II Item 1A. Risk Factors of this Quarterly Report on this Form 10-Q and Part I Item 1A. Risk Factors of our Annual Report on Form 10-K filed with the Securities and Exchange Commission (SEC) on March 24, 2022 for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Quarterly Report on Form 10-Q will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame or at all. We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
You should read this Quarterly Report on Form 10-Q and the documents that we reference in this Quarterly Report on Form 10-Q and have filed as exhibits to this Quarterly Report on Form 10-Q completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations should be read in conjunction with: (i) the interim consolidated financial statements and related notes thereto which are included in this Quarterly Report on Form 10-Q; and (ii) our annual financial statements for the year ended December 31, 2021 which are included in our Annual Report on Form 10-K filed with the SEC on March 24, 2022.
Overview
We are a pharmaceutical company focused on the development and commercialization of products for patients suffering from rare genetic epilepsies and other seizure disorders. On March 18, 2022, the FDA approved our new drug application (NDA) for the use of ZTALMY® (ganaxolone) oral suspension for the treatment of seizures associated with CDD in patients 2 years of age and older. This is our first FDA-approved product which we plan to commercialize in the United States. We also plan to continue to develop ganaxolone for other rare genetic epilepsies and for a number of additional indications including other seizure disorders. While the precise mechanism by which ganaxolone exerts its therapeutic effects in the treatment of seizures associated with CDD is unknown, its anticonvulsant effects are thought to result from positive allosteric modulation of the gamma-aminobutyric acid type A (GABAA) receptor in the Central Nervous System (CNS). Ganaxolone is being developed in formulations for two different routes of administration: intravenous (IV) and oral. Ganaxolone is a synthetic analog of allopregnanolone, an endogenous neurosteroid. The different formulations are intended to maximize potential therapeutic applications of ganaxolone for adult and pediatric patient populations, in both acute and chronic care, and for both in-patient and self-administered settings. Ganaxolone acts at both synaptic and extrasynaptic GABAA receptors, a target known for its anti-seizure, antidepressant and anxiolytic potential.
COVID-19
The continued global spread of COVID-19, including the Omicron variant and its subvariants, has impacted our clinical operations and timelines. For example, our Phase 3 RAISE trial in RSE is conducted in hospitals, including intensive care units and academic medical centers, which have experienced high rates of COVID-19 admissions. Several academic medical centers and intensive care units participating in the RAISE trial have experienced COVID-related difficulties, including staff turnover and the need to devote significant resources to patients with COVID-19, which has resulted in site initiation and enrollment delays for the RAISE trial. Given these COVID-19-related challenges and our recent interruption in drug supply, we now expect our top-line data readout for the RAISE trial to be available in the second half of 2023. In May 2022, we resumed screening and recruitment for the RAISE trial. In addition, our ganaxolone clinical trials in the outpatient setting may be negatively impacted if patients and their caregivers do not want
22
to participate while the COVID-19 pandemic persists. The duration and severity of the pandemic and its long-term impact on our business are uncertain at this time.
Our Products and Product Candidates
ZTALMY® (ganaxolone) oral suspension CXX
ZTALMY is an oral suspension given three times per day that we have developed for the treatment of CDD. ZTALMY was approved by the FDA in March 2022 for the treatment of seizures associated with CDD in patients 2 years of age and older. ZTALMY is pending scheduling by the DEA as a controlled substance under the CSA, which we expect to be completed in June 2022 (DEA Scheduling). We expect to have ZTALMY available for commercial sale and shipment to patients with a prescription in the U.S. in mid-July 2022.
CDD is a serious and rare genetic disorder that is caused by a mutation of the CDKL5 gene, located on the X chromosome. CDD is a severely debilitating and potentially fatal genetic condition, which occurs with an estimated frequency of 1:40,000 live births in the U.S. It predominantly affects females and is characterized by early onset, difficult to control seizures and severe neurodevelopmental impairment. The CDKL5 gene encodes proteins essential for normal brain function. Most children affected by CDD have neurodevelopmental deficits such as difficulty walking, talking and taking care of themselves. Many also suffer from scoliosis, gastrointestinal dysfunction or sleep disorders. Genetic testing is available to determine if a patient has a mutation in the CDKL5 gene.
In June 2017, we were granted FDA orphan drug designation for ganaxolone for the treatment of CDD. Additionally, in November 2019, the European Medical Agency’s (EMA) Committee for Orphan Medicinal Products (COMP) granted orphan drug designation for ganaxolone for the treatment of CDD. In July 2020, the FDA granted Rare Pediatric Disease Designation (RPD Designation) for ganaxolone for the treatment of CDD. The FDA grants RPD Designation for diseases that affect fewer than 200,000 people in the U.S. and in which the serious or life-threatening manifestations occur primarily in individuals 18 years of age and younger. The approval of ZTALMY in CDD is based on data from the Phase 3 Marigold double-blind placebo-controlled trial, in which 101 patients were randomized and treated with ZTALMY. Patients showed a median 30.7% reduction in 28-day major motor seizure frequency, compared to a median 6.9% reduction for those receiving placebo, achieving the trial’s primary endpoint (p=0.0036). In the Marigold open label extension study, patients treated with ZTALMY for at least 12 months (n=48) experienced a median 49.6% reduction in major motor seizure frequency. In the clinical development program, ZTALMY demonstrated efficacy, safety and tolerability with the most common adverse reactions (AEs) (incidence >5% and at least twice the rate of placebo) in the ZTALMY group being somnolence, pyrexia, salivary hypersecretion, and seasonal allergy.
Priority Review Voucher. As a result of the RPD Designation for ganaxolone for the treatment of CDD, the FDA awarded us a Rare Pediatric Disease Priority Review Voucher (PRV). This voucher may be redeemed for priority review by the FDA in a subsequent marketing application. Vouchers can be sold or transferred to a third party by the voucher recipient. We intend to monetize the PRV to fund on-going operations, including continued clinical development and commercialization efforts for ganaxolone.
Commercial Strategy. Since ZTALMY was approved by the FDA, we are focused on implementing and executing an integrated launch plan making ZTALMY available upon DEA Scheduling to CDD patients through a specialty pharmacy. Key launch strategies include: (1) establishing our supply chain network and quality management system to assure product is available to patients; (2) driving clinical awareness of ZTALMY as the first and only FDA approved product indicated specifically for seizures associated with CDD; (3) deploying our field sales force to target physicians who treat this rare pediatric patient population; (4) engaging commercial and government payers with the objective of obtaining insurance coverage; and (5) developing our internal capabilities (such as Finance, Human Resources, Information Technology, Data Analytics and Compliance) to support our first launch as a commercial company.
Marketing Strategy. At launch, our strategy is to reinforce that seizures are central to the constellation of CDD symptoms, establish ZTALMY as central to the comprehensive management of CDD, and ensure that patients have
23
seamless access to ZTALMY from prescription through fulfillment. Our “Now Approved” marketing campaign for ZTALMY is now live, and our integrated commercial launch activities are on track to begin in July 2022.
Sales Strategy. Our sales leadership is in place, and we have recently hired 16 regional account managers experienced in rare disease as our commercial sales force. Our field force will target identified key accounts and centers of excellence for CDD. Based on our market research, we estimate the addressable patient population for ZTALMY in CDD in the U.S. is approximately 2,000 patients. As this is the first product approved by the FDA specifically for seizures associated with CDD and the International Classification of Diseases, Tenth Revision (ICD10) code for CDD was established in 2020, there is limited data available for this specific market. We have strengthened both our market access and field force teams, and both payer and customer engagement are underway.
Market Access. We have established a cross-functional payer and reimbursement account team with the objective of obtaining and maintaining reimbursement (coverage) of ZTALMY. We plan to focus our efforts on reimbursement from commercial payers where pharmacy benefit managers (PBMs) control the majority of commercial pharmacy-benefit lives and government payers, primarily Medicaid for the target population for CDD. We expect approximately 60% of the CDD patient population will access coverage through both Fee-for-Service and Managed Medicaid, with the remaining 40% accessing commercial coverage, with the top PBMs having significant influence. Preparations continue to advance for a simultaneous launch of The ZTALMY One™ Program, a patient services program to provide assistance to healthcare providers, patients and caregivers with product access and ongoing product support, and offers programs for eligible patients who need financial support for their ZTALMY prescription.
Specialty Pharmacy. We have engaged a specialty pharmacy to provide services for patients, including patient enrollment, benefit verification and investigation, prior authorization support, patient education and drug counseling, dispensing of product and shipment coordination.
Infrastructure. We continue to develop our internal capabilities and processes to support a commercial stage company. We have implemented a healthcare compliance program to guide our compliance with rules and regulations regarding pharmaceutical sales.
Manufacture of Commercial Supply. We are in the process of negotiating a commercial supply agreement for ganaxolone active pharmaceutical ingredient with our current manufacturer. We are also finalizing a commercial supply agreement with our current supplier of finished bulk drug product.
Regulated as a Controlled Substance. ZTALMY is expected to be regulated by the DEA as a controlled substance under the CSA. Under the CSA, drugs are classified into five (5) distinct categories or schedules depending upon the drug’s acceptable medical use and the drug’s abuse or dependency potential. Based on our assessment of the abuse-related preclinical and clinical data on ganaxolone and its mechanism of action, we expect ZTALMY to be a Schedule IV drug. Schedule IV is defined by the DEA as drugs with a low potential for abuse and low risk of dependence. We expect this scheduling process to take approximately 90 days. We plan to commercially launch ZTALMY after this scheduling process is completed. As a controlled substance, ganaxolone will be subject to the applicable CSA requirements such as registration, security, recordkeeping and reporting, storage manufacturing, distribution, importation and other requirements.
Post-Marketing Requirements. In connection with the FDA approval of ZTALMY for CDD, we have a number of post-marketing commitments which include: 2-year carcinogenicity studies of ganaxolone and the major human unconjugated plasma metabolite, M2, in rats; a 26-week carcinogenicity of ganaxolone in transgenic mice; a juvenile animal toxicity study of the major human unconjugated plasma metabolite, M2, in rats; Phase 1 renal and hepatic impairment studies and a thorough QTc study; extractable/leachable study results on the container closure system; a CNS distribution study of the M47 metabolite in rats; and in vitro studies to assess the drug interaction potential of M47 metabolite. The Phase 1 renal impairment study was completed and submitted to the FDA in May 2022. We expect to be able to complete the remaining required FDA studies within the requested FDA timeframe.
24
Marketing Authorization Application
In August 2021, the Committee for Medicinal Products for Human Use (CHMP) of the EMA granted our request for accelerated assessment of ganaxolone for the treatment of seizures associated with CDD. The marketing authorization application (MAA) for ganaxolone was submitted to the EMA on October 11, 2021 and on October 28, 2021 we received formal notification from the EMA that the CDD MAA was validated. With this validation, the EMA began its formal review of the MAA under the centralized procedure for all member states of the EU, Norway, Iceland, and Liechtenstein.
In February 2022, the MAA was converted to a standard review and we reached an agreement with the EMA to extend the Day 120 clock stop by three months to allow sufficient time to respond to questions received as part of the review process. As a result, we are targeting submission of responses to the EMA by mid-year and expect the CHMP’s opinion on the MAA by the end of 2022. Further delays in the review and approval process could occur if we are not able to timely or adequately respond to all EMA requests or if the EMA does not agree that our responses are adequate to address its questions.
Our Pipeline
We are developing ganaxolone in indications where there is a mechanistic rationale for ganaxolone to provide a benefit, including the following indications:
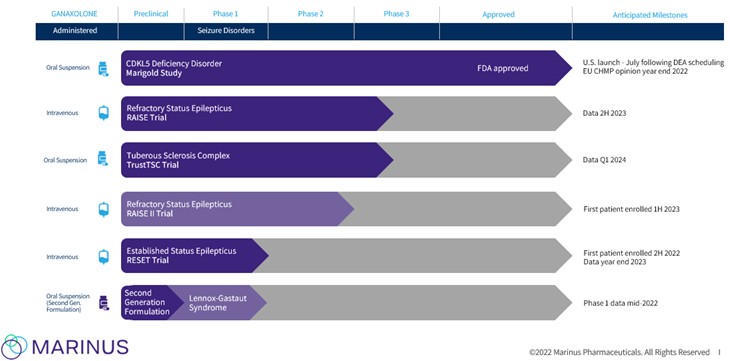
Status Epilepticus (SE)
SE is a life-threatening condition characterized by continuous, prolonged seizures or rapidly recurring seizures without intervening recovery of consciousness. If SE is not treated urgently, permanent neuronal damage may occur, which contributes to high rates of morbidity and mortality. Patients with SE who do not respond to first-line benzodiazepine treatment are classified as having ESE and those who then progress to and then fail at least one second-line antiepileptic drug (AED) are classified as having RSE. In RSE, synaptic GABAA receptors are internalized into the neuron, resulting in decreased responsiveness to drugs such as benzodiazepines. Current treatment for RSE unresponsive to one or more second-line AEDs is IV anesthesia to terminate seizures and prevent neuronal injury and other complications. The IV anesthetic is increased to a level that induces deep coma and is maintained at that rate for 24 hours or more. SE that recurs following an attempted wean of IV anesthesia is classified as super refractory status epilepticus
25
(SRSE). In April 2016, we were granted FDA orphan drug designation for IV formulation of ganaxolone for the treatment of SE, which includes RSE.
We own a family of pending patent applications that claim certain therapeutic regimens for the treatment of SE, including RSE, using intravenous ganaxolone. In September 2021, the United States Patent Office granted us a patent on a method of treating SE, including dosing regimens. This issued patent expires in 2040.
In January 2021, we enrolled the first patient in the Phase 3 pivotal RAISE trial. The RAISE trial is a randomized, double-blind, placebo-controlled clinical trial in patients with RSE. We expect approximately 80 trial sites in hospitals, primarily across the U.S. and Canada, to participate. The RAISE trial is designed to enroll approximately 124 patients, who will be randomized to receive ganaxolone or placebo added to standard of care second-line AED. With this number of patients, the trial is designed to provide over 90% power to detect a 30% efficacy difference between ganaxolone and placebo.
The co-primary endpoints for the RAISE trial are (1) proportion of patients with RSE who experience seizure cessation within 30 minutes of treatment initiation without other medications for RSE treatment, and (2) proportion of patients with no progression to IV anesthesia for 36 hours following initiation of the study drug. Several academic medical centers and intensive care units participating in the RAISE trial have experienced COVID-related difficulties, including staff turnover and the need to devote significant resources to patients with COVID-19, which has resulted in site initiation and enrollment delays for the RAISE trial. Additionally, in February 2022, we temporarily paused the RAISE trial after routine monitoring of stability batches of clinical supply material indicated that it became necessary to reduce the shelf life to less than the anticipated 24-months to meet product stability testing specifications. We notified the FDA of this issue and our plans to proactively pause the trial, and we subsequently provided additional information to the FDA to support resuming trial activities. In May 2022, we announced that the trial has resumed utilizing new batches of the current IV formulation of ganaxolone. We have implemented a reduced shelf life of 12 months. In agreement with the FDA, ganaxolone clinical supplies will be stored under refrigerated conditions for the entire duration of clinical use. We anticipate modifying the IV ganaxolone formulation with a new buffer by end of the third quarter of 2022, targeting a shelf life of at least 24 months. We continue to expand the number of participating clinical trial sites for the RAISE trial in the U.S. and Canada, and are now expanding target sites to include Israel and Australia sites as well, and are working closely with key investigators and site coordinators to support enrollment efficiencies with more than 50 sites now activated. Consistent with the prior announcement, we expect our top-line data readout for the RAISE trial to be available in the second half of 2023.
Planning continues for a separate RSE trial to support a MAA in Europe (RAISE II trial). Following a meeting with the EMA in the first quarter of 2021, at which we discussed trial design, and due to the delay in clinical trial supply mentioned for the RAISE trial, the RAISE II trial initiation is planned for the first half of 2023. The RAISE II trial will be a double blind, placebo-controlled pivotal registration trial expected to enroll 70 patients who have failed first-line benzodiazepine treatment and at least one prior second-line AED. Patients will receive either ganaxolone or placebo, administered in combination with a standard-of-care second-line AED. The RAISE II trial will provide data that is complementary to the RAISE trial, with ganaxolone or placebo being administered in combination with a standard-of-care AED. There are two additional key differences between the RAISE trial and the RAISE II trial. First, rather than including only progression to IV anesthesia as a treatment failure, the endpoint for RAISE II will include any escalation of care. This could be IV anesthesia or another second-line IV AED. Second, the primary analysis for the RAISE II trial will be a responder analysis, with response defined as SE cessation within 30 minutes and no escalation of care within 36 hours. The RAISE trial specifies a co-primary endpoint, requiring statistical significance for both early onset and durability of effect.
The FDA has indicated alignment on the overall trial design for a third SE study, the RESET trial, which will study ganaxolone in ESE. We are making preparations to begin U.S. enrollment of this Phase 2 clinical trial in the second half of 2022. The RESET trial is expected to be conducted in emergency rooms under exception from informed consent guidelines and is expected to enroll patients with convulsive ESE. We expect that the RESET trial will be composed of two stages, the initial open-label, dose optimization stage and subsequent double-blind placebo-controlled stage. We anticipate that during the open-label portion of the trial, multiple sequential cohorts of patients will be assessed to determine the bolus dose and subsequent IV infusion rate and infusion duration to be used in the double-blind second
26
stage of the trial. We expect that this double-blind placebo-controlled phase will enroll approximately 80 ESE patients equally distributed between two arms of the trial who, in addition to standard of care, will receive either IV ganaxolone or placebo. We also expect that the primary efficacy endpoint of the RESET trial will be the absence of electrographic (rapid EEG) evidence of SE or recurrence of generalized convulsions at 1 hour after the initiation of treatment. We are targeting data from the first dose-finding cohorts of the RESET trial by the end of 2023.
Tuberous Sclerosis Complex (TSC)
TSC is a rare genetic disorder that affects many organs and causes non-malignant tumors in the brain, skin, kidney, heart, eyes, and lungs. The condition is caused by inherited mutations in either the TSC1 gene or the TSC2 gene. TSC occurs with a frequency of 1:6,000 live births and a mutation is found in 85% of patients. While the disease phenotype can be extremely variable, epilepsy occurs in up to 85% of TSC patients. TSC is a leading cause of genetic epilepsy, often manifesting in the first year of life as either focal seizures or infantile spasms. There are currently few disease-specific treatments approved for seizures in TSC. Orphan drug designation for ganaxolone for the treatment in TSC was granted by the FDA in August 2021 and by the EMA in October 2021.
In August 2021, we announced top-line data from our open-label Phase 2 trial (CALM trial) evaluating the safety and efficacy of adjunctive oral ganaxolone in 23 patients with seizures associated with TSC. The CALM trial enrolled 23 patients ages 2 to 32 who entered a four-week baseline period followed by a 12-week treatment period, during which they received up to 600 mg of ganaxolone (oral liquid suspension) three times a day. Patients who met eligibility criteria were able to continue ganaxolone treatment during a 24-week extension. The primary endpoint was the percent change in 28-day TSC-associated seizure frequency during the 12-week treatment period relative to the four-week baseline period. Secondary outcome measures included the percentage of patients experiencing a greater than or equal to 50% reduction in 28-day TSC-associated seizure frequency through the end of the 12-week treatment period compared to the 4-week baseline period.
The primary endpoint showed a median 16.6% reduction in 28-day frequency of TSC-associated seizures relative to the four-week baseline period. A secondary endpoint showed that the proportion of patients that achieved at least a 50% seizure reduction was 30.4%. During the trial, patients with focal seizures (n=19) showed a median 25.2% reduction in focal seizure frequency. Ganaxolone was generally well-tolerated with somnolence reported as the most common AE. In addition, one serious adverse event (SAE) of worsening seizures occurred, which was assessed by the investigator as treatment-related. Four patients discontinued the trial due to AEs. Additionally, the data from the trial suggested that in patients on concomitant Epidiolex, early elevation of ganaxolone blood levels occurred and appeared to be linked to greater somnolence. We have adjusted the titration schedule in the Phase 3 TSC trial, with the goal of improving tolerability.
In response to our request for an End of Phase 2 meeting with the FDA regarding a proposed Phase 3 TSC trial, the FDA provided written responses to our questions in lieu of a meeting. We believe the written responses show overall alignment on the clinical development plan in TSC. We also believe that, based on the FDA’s written responses, and with the FDA approval of CDD, a single trial could serve the necessary support for an NDA for TSC in the U.S. In response to our request for Protocol Assistance, which is a special form of scientific advice available for developers of designated orphan medicines for rare diseases, the EMA provided written feedback in December 2021 in lieu of a meeting. We believe the written responses from the EMA, similar to those from the FDA, show overall alignment on the clinical development plan in TSC. After commencing site initiations in the first quarter of 2022, we are actively screening patients in the U.S. for enrollment in a global Phase 3 randomized, double blind, placebo-controlled trial (TrustTSC trial) of adjunctive ganaxolone in approximately 160 TSC patients. We expect to expand trial to include a target of 65-75 sites, including several TSC centers of excellence, predominantly in the U.S., Western Europe, Canada and Israel. The primary endpoint for the TrustTSC trial is percent change in 28-day frequency of TSC-associated seizures. We plan to announce top-line data from the TrustTSC trial in the first quarter of 2024.
Lennox-Gastaut Syndrome (LGS)
LGS is a severe form of epilepsy that typically begins between one and eight years of age. Affected children experience multiple seizure types that are often unresponsive to treatment, the most common being atonic, tonic and atypical absence seizures. Children with LGS may also have neurodevelopmental delay and behavioral problems.
27
We plan to pursue the development of ganaxolone for LGS, given the overlap in seizure types and etiologies with other disorders where ganaxolone has potential to improve clinical outcomes, such as CDD and TSC. We are planning to utilize a second-generation formulation of ganaxolone for our LGS development program. We expect top-line data from our Phase 1 trial with healthy volunteers utilizing a second-generation formulation of ganaxolone to be available mid-2022. Provided that evaluation of the Phase 1 trial data supports moving to a Phase 2 clinical trial, we plan to begin a Phase 2 LGS clinical trial in the second half of 2022. A second formulation candidate which we believe has potential for an improved pharmacokinetic profile and reduced dosing frequency has been selected, and we plan to begin Phase 1 development of this product candidate in the first quarter of 2023.
Operations
Our operations to date have consisted primarily of organizing and staffing our company and developing ganaxolone, including conducting preclinical studies, clinical trials and raising capital. We have funded our operations primarily through sales of equity and debt securities. We have no products currently available for sale, have incurred operating losses since inception, have not generated any product sales revenue and have not achieved profitable operations. We incurred a net loss of $19.4 million and $27.1 million for the three months ended March 31, 2022 and 2021, respectively. Our accumulated deficit as of March 31, 2022 was $430.1 million, and we expect to continue to incur substantial losses in future periods. We anticipate that our operating expenses will increase substantially as we carry out all of our planned commercialization and continued research and development activities with respect to ganaxolone.
We anticipate that our expenses will increase substantially as we:
● | conduct multiple later stage clinical trials in targeted indications; |
| ● | continue the research, development and scale-up manufacturing capabilities to optimize ganaxolone and dose forms for which we may obtain regulatory approval; |
| ● | establish and implement sales, marketing and distribution capabilities to commercialize ganaxolone; |
| ● | conduct other preclinical studies and clinical trials to support the filing of NDAs with the FDA, MAAs with the EMA and other marketing authorization filings with regulatory agencies in other countries; |
| ● | acquire the rights to other product candidates and fund their development; |
| ● | maintain, expand and protect our global intellectual property portfolio; |
| ● | hire additional clinical, manufacturing, scientific and commercial personnel; and |
| ● | add operational, financial and management information systems and personnel, including personnel to support our drug development efforts. |
We had cash and cash equivalents of $126.3 million at March 31, 2022. We believe that our existing cash and cash equivalents on hand as of March 31, 2022 will be sufficient to fund our operating expenses, capital expenditure requirements and maintain the minimum cash balance required under our debt facility into the first quarter of 2023. However, we will need to secure additional funding in the future, from one or more equity or debt financings, government funding, collaborations, licensing transactions, other commercial transactions or other sources, such as the monetization of our Priority Review Voucher, in order to carry out all of our commercialization and planned research and development activities with respect to ganaxolone.
28
Financial Overview
Federal Contract Revenue
In September 2020, we and BARDA entered into the BARDA Contract, under which we received an award of up to an estimated $51 million for development of IV-administered ganaxolone for the treatment of RSE. The BARDA Contract provides for funding to support, on a cost-sharing basis, the completion of a Phase 3 clinical trial of IV-administered ganaxolone in patients with RSE, which covers the RAISE trial, funding of pre-clinical studies to evaluate IV-administered ganaxolone as an effective treatment for RSE due to chemical nerve gas agent exposure, and funding of certain ganaxolone manufacturing scale-up and regulatory activities. In March 2022, we entered into an amendment with BARDA to extend the end date of our base performance period for funding under the BARDA Contract from September 1, 2022 to December 31, 2023.
The BARDA Contract consists of an approximately two-year base period, and an extension period through December 31, 2023 per the amendment described above, during which BARDA will provide up to approximately $21 million of funding for the RAISE trial on a cost share basis and funding of additional preclinical studies of ganaxolone in nerve agent exposure models. Following successful completion of the RAISE trial and preclinical studies in the base period and extension period, the BARDA Contract provides for approximately $30 million of additional BARDA funding for three options in support of ganaxolone manufacturing, supply chain, clinical, regulatory and toxicology activities. Under the BARDA Contract, we will be responsible for cost sharing in the amount of approximately $33 million and BARDA will be responsible for approximately $51 million, if all development options are completed. The contract period-of-performance (base period plus option exercises) is up to approximately five years.
We recognize federal contract revenue from the BARDA Contract in the period in which the allowable research and development expenses are incurred. We expect federal contract revenue to increase as the costs associated with our RAISE trial increase.
Collaboration Revenue
In July 2021, we and Orion entered into a collaboration agreement (Orion Collaboration Agreement). Under the terms of the Orion Collaboration Agreement, we granted Orion an exclusive, royalty-bearing, sublicensable license to certain of our intellectual property rights with respect to commercializing biopharmaceutical products incorporating ganaxolone (Licensed Products) in the European Economic Area, the United Kingdom and Switzerland (collectively, the Territory) for the diagnosis, prevention and treatment of certain human diseases, disorders or conditions (Field), initially in the indications of CDD, TSC and RSE.
Under the terms of the Orion Collaboration Agreement, we received a €25.0 million ($29.6 million) upfront payment from Orion in July 2021. In connection with the upfront fee, we agreed to provide Orion with the results of a planned genotoxicity study on the M2 metabolite of ganaxolone, a “Combined Micronucleus & Comet study in vivo.” In the event that the results of such study were positive, based on the criteria set forth in the study’s protocol, Orion would have had the right to terminate the Orion Collaboration Agreement within ninety (90) days after its receipt of the final report of such study, in which case we would have been required to refund Orion seventy-five percent (75%) of the upfront fee. In the event of such termination and refund, Orion would have had no further rights pursuant to the oral and IV dose formulations of ganaxolone and the Orion Collaboration Agreement would have terminated and be of no further force or effect. In February 2022, the verified draft study report showed that no genotoxicity was found, as measured by formation of micronuclei in the bone marrow or comet morphology in the liver. These results were formalized in the final study report received in May 2022 and, as a result of the study’s findings, Orion does not have the right to terminate the Orion Collaboration Agreement based on the study outcome. We are eligible to receive up to an additional €97.0 million in research and development reimbursement and cash milestone payments based on specific clinical and commercial achievements, as well as tiered royalty payments based on net sales ranging from the low double-digits to high teens for the oral programs and the low double-digits to low 20s for the IV program. Also, as part of the overall arrangement, we have agreed to supply the Licensed Products to Orion at an agreed upon price.
29
We identified the following commitments under the arrangement: (i) exclusive rights to develop, use, sell, have sold, offer for sale and import any product comprised of Licensed Product (License); (ii) development and regulatory activities (Development and Regulatory Activities); and (iii) requirement to supply Orion with the Licensed Product at an agreed upon price (Supply of Licensed Product). We determined that these three commitments represent distinct performance obligations for purposes of recognizing revenue and will recognize collaboration revenue or a reduction of expense as we fulfill each performance obligation.
Research and Development Expenses
Our research and development expenses consist primarily of costs incurred for the development of ganaxolone, which include:
● | employee-related expenses, including salaries, benefits, travel and stock-based compensation expense; |
● | expenses incurred under agreements with clinical research organizations (CROs) and investigative sites that conduct our clinical trials and preclinical studies; |
● | the cost of acquiring, developing and manufacturing clinical trial materials; |
● | facilities, depreciation and other expenses, which include direct and allocated expenses for rent and maintenance of facilities, insurance and other supplies; |
● | costs associated with preclinical activities and regulatory operations; and |
● | costs associated with developing new formulations and prodrugs of ganaxolone. |
We expense research and development costs when we incur them. We record costs for some development activities, such as clinical trials, based on an evaluation of the progress to completion of specific tasks using data such as patient enrollment, clinical site activations and information our vendors provide to us.
We have and will incur substantial costs beyond our present and planned clinical trials in order to file an NDA and Supplemental NDAs, or MAAs outside the U.S., for ganaxolone for various clinical indications, and in each case, the nature, design, size and cost of further clinical trials and other studies will depend in large part on the outcome of preceding studies and trials and discussions with regulators. It is difficult to determine with certainty the costs and duration of our current or future clinical trials and preclinical studies, or if, when or to what extent we will generate revenue from the commercialization and sale of ganaxolone if we obtain regulatory approval. We may never succeed in achieving regulatory approval for ganaxolone. The duration, costs and timing of clinical trials and development of ganaxolone will depend on a variety of factors, including the uncertainties of future clinical trials and preclinical studies, uncertainties in clinical trial enrollment rate and significant and changing government regulation.
In addition, the probability of success for our clinical programs will depend on numerous factors, including competition, manufacturing capability and commercial viability. Our commercial success depends upon attaining significant market acceptance, if approved, among physicians, patients, healthcare payers and the medical community. We will determine which programs to pursue and how much to fund each program in response to the scientific and clinical success, as well as an assessment of commercial potential.
IP License Fee Expenses
In March 2022, we entered into an exclusive patent license agreement (License Agreement) with Ovid Therapeutics Inc. (Ovid). Under the License Agreement, we have an exclusive, non-transferable (except as provided in the License Agreement), royalty-bearing, sublicensable license under certain of Ovid’s patent(s) and patent applications to develop, make, have made, commercialize, promote, distribute, sell, offer for sale and import, ganaxolone, including any analogues or derivatives, including its salts, and pharmaceutical formulations of the foregoing (Licensed Products),
30
in the U.S., the member states of the EU, Iceland, Lichtenstein, Norway, the United Kingdom, and Switzerland (Territory) for the treatment of CDD in humans (Field). Under the License Agreement, we have the sole right and responsibility for, and control over, all development, manufacturing, and commercialization activities, including all regulatory activities, with respect to the Licensed Products in the Field in the Territory. In addition, all regulatory approvals and related filings with respect to the Licensed Products in the Field in the Territory will be in the name of, and be owned solely by, us. We were required, at Ovid’s option exercisable in accordance with the License Agreement, to (i) pay to Ovid the sum of $1.5 million in cash; or (ii) issue to Ovid 123,255 shares of our common stock, which option to obtain shares of our common stock was exercisable within the five-business day period following the filing of our Annual Report on Form 10-K for the year ended December 31, 2021 on March 24, 2022. On March 29, 2022, we issued 123,255 shares of our common stock to Ovid, per Ovid’s option in accordance with the License Agreement. As such, we recorded $1.2 million of IP license fee expenses related to the Ovid License Agreement in the three months ended March 31, 2022.
The License Agreement also provides for payment of royalties by us to Ovid in the low single digits on net sales by us, our affiliates and sublicensees, of Licensed Products in the Field in the Territory. Such royalties are subject to reduction in the event of generic competition in accordance with the License Agreement. We may terminate the License Agreement at any time without cause on thirty days’ prior written notice. Either party may terminate the License Agreement for the other party’s material breach or insolvency subject to certain cure periods. Also, Ovid has the right to terminate the License Agreement if there has not been a first commercial sale of any Licensed Products in the Field in the Territory on or before June 30, 2025. In the event of termination, all licenses granted under the License Agreement will terminate.
General and Administrative Expenses
General and administrative expenses consist principally of salaries and related costs for executive and other administrative personnel and consultants, including stock-based compensation and travel expenses. Other general and administrative expenses include professional fees for legal, patent review, consulting and accounting services. General and administrative expenses are expensed when incurred. We expect that our general and administrative expenses will increase in the future as a result of employee hiring and our scaling-up of operations commensurate with supporting more advanced clinical trials and for commercial infrastructure. These increases will likely include increased costs for insurance, hiring of additional personnel, outside consultants, and legal counsel and accountants, among other expenses.
Interest Income
Interest income consists principally of interest income earned on cash and cash equivalents and investment balances.
Interest Expense
Interest expense consists of interest expense, amortization of debt discount and commitment fees related to our Notes Payable.
Other Income (Expense)
Other income and expense consists principally of gains or losses on disposal of fixed assets held for sale, foreign currency translation, and fair value adjustments.
Results of Operations
Federal Contract Revenue
We recognized $1.5 million and $1.8 million of federal contract revenue for the three months ended March 31, 2022 and 2021, respectively, as a result of the BARDA Contract.
31
Collaboration Revenue
Collaboration revenue was $12.7 million for the three months ended March 31, 2022, as a result of revenue recognition related to the previously refundable upfront payment pursuant to the Orion Collaboration Agreement. In connection with the upfront fee, we agreed to provide Orion with the results of an ongoing genotoxicity study. In February 2022, the verified draft study report showed that no genotoxicity was found, as measured by formation of micronuclei in the bone marrow or comet morphology in the liver. These results were formalized in the final study report received in May 2022 and, as a result of the study’s findings, we are not required to refund Orion any of the upfront fee and Orion does not have the right to terminate the Orion Collaboration Agreement based on the study outcome. During the three months ended March 31, 2022, we allocated the previously refundable portion of the upfront payment to the transaction price and recognized the related revenue. We did not recognize any collaboration revenue for the three months ended March 31, 2021.
Research and Development Expenses
We allocate direct research and development expenses, consisting principally of external costs, such as fees paid to investigators, consultants, central laboratories and CROs in connection with our clinical trials, and costs related to manufacturing, to specific product development programs. We do not allocate costs related to purchasing clinical trial materials, employee and contractor-related costs, costs associated with our facility expenses, including depreciation or other indirect costs, to specific product programs because these costs are deployed across multiple product programs under research and development and, as such, are separately classified. The table below shows our research and development expenses incurred with respect to each active program, in thousands. The primary drivers of our research and development expenditures are currently in our product development programs in TSE, RSE, TSC and PCDH19. We expect our research and development expenses for ganaxolone will continue to increase during subsequent periods. We did not allocate research and development expenses to any other specific product development programs during the periods presented (in thousands):
Three Months Ended | |||||||
March 31, | |||||||
2022 | 2021 | ||||||
CDKL5 deficiency disorder (1) |
| $ | 855 |
| $ | 1,839 |
|
PCDH19-related epilepsy (2) | 580 | 998 | |||||
Tuberous Sclerosis (3) | 1,593 | 859 | |||||
Drug Development - Suspension (4) | 1,348 | 1,838 | |||||
Oral Indications Subtotal | 4,376 | 5,534 | |||||
Status epilepticus (5) | 1,869 | 1,365 | |||||
Drug Development - IV | 1,708 | 1,738 | |||||
IV Indications Subtotal | 3,577 | 3,103 | |||||
Other research and development (6) | 1,447 | 3,135 | |||||
Indirect research and development (7) | 8,591 | 6,819 | |||||
Total | $ | 17,991 | $ | 18,591 | |||
Note: Certain prior year expenses have been reclassified to conform to current year presentation.
| (1) | The decrease in the three months ended March 31, 2022 compared to the 2021 period was due primarily to more significant regulatory and statistical analysis expenses associated with our NDA filing preparation in the prior period and reduced clinical trial activity in the current period. |
| (2) | The decrease in the three months ended March 31, 2022 compared to the 2021 period was due primarily to lower current period costs associated with our on-going open label extension. |
32
| (3) | The increase in the three months ended March 31, 2022 compared to the 2021 period was due primarily to increased activity in the 2022 period from the Phase 3 TSC trial start-up, as compared to more limited Phase 2 activities in the relevant 2021 period. |
| (4) | The decrease in the three months ended March 31, 2022 compared to the 2021 period was due primarily to higher manufacturing costs related to pre-validation and registration batches in the prior period compared to the relevant 2022 period. |
| (5) | The increase in the three months ended March 31, 2022 compared to the 2021 period was due primarily to increased costs related to the RESET trial, with no comparable costs in the relevant 2021 period. |
| (6) | Other research and development expenses include external expenses associated with preclinical and clinical development of ganaxolone, including safety studies, stability studies, preclinical studies, including animal toxicology and pharmacology studies, and other professional fees. The decrease in the three months ended March 30, 2022 compared to the 2021 period was due primarily to reduced level of safety study activities. |
| (7) | The increase in the three months ended March 31, 2022 compared to the 2021 period was related to increased personnel costs in support of our increased activity in preclinical, clinical, and manufacturing activities. |
General and Administrative Expenses
General and administrative expenses were $11.7 million for the three months ended March 31, 2022, compared to $10.4 million for the three months ended March 31, 2021. The primary drivers of the increase for the three months ended March 31, 2022 were $1.8 million in increased personnel costs, $0.6 million in increased consulting costs, $0.4 million in increased commercialization preparation and $0.2 million in increased software related expenses, partially offset by $1.7 million decreased noncash stock-based compensation costs. Of the decreased noncash stock-based compensation costs, $2.1 million was due to modifications of stock options recorded in the first quarter of 2021 in connection with a severance agreement with our former Chief Financial Officer.
Interest Expense
Interest expense was $1.7 million for the three months ended March 31, 2022, inclusive of $1.3 million of interest paid, $0.3 million of debt amortization, and less than $0.1 million related to commitment fees accrued in the three months ended March 31, 2022 in connection with our Notes payable (Note 8 in accompanying notes to consolidated financial statements). No interest expense was recorded for the three months ended March 31, 2021.
Liquidity and Capital Resources
Since inception, we have incurred net losses and negative cash flows from our operations. We incurred net losses of $19.4 million and $27.1 million for the three months ended March 31, 2022 and 2021, respectively. Our cash used in operating activities was $27.7 million for the three months ended March 31, 2022 compared to $16.2 million for the three months ended March 31, 2021. Historically, we have financed our operations principally through the sale of common stock, notes payable, preferred stock and convertible debt. At March 31, 2022, we had cash and cash equivalents of $126.3 million.
European Commercialization Agreement
On July 30, 2021, we entered into the Orion Collaboration Agreement, whereby Orion received exclusive rights to commercialize the oral and IV dose formulations of ganaxolone in the European Economic Area, United Kingdom and Switzerland in multiple seizure disorders, including CDD, TSC and RSE. Under the agreement, we received a €25 million ($29.6 million) upfront fee and are eligible to receive up to an additional €97 million in research and development reimbursement and cash milestone payments based on specific clinical and commercial achievements, as well as tiered royalty payments based on net sales ranging from the low double digits to the high teens for the oral programs and the low double-digits to the low twenties for the IV programs. In connection with the upfront fee, we
33
agreed to provide Orion with the results of an ongoing genotoxicity study on the M2 metabolite of ganaxolone, a “Combined Micronucleus & Comet study in vivo.” In the event that the results of such study were positive, based on the criteria set forth in the study’s protocol, Orion would have had the right to terminate the Orion Collaboration Agreement within ninety (90) days after its receipt of the final report of such study, in which case we would have been required to refund Orion seventy-five percent (75%) of the upfront fee. In the event of such termination and refund, Orion would have had no further rights pursuant to the oral and IV dose formulations of ganaxolone and the Orion Collaboration Agreement would have terminated and been of no further force or effect. In February 2022, the verified draft study report showed that no genotoxicity was found, as measured by formation of micronuclei in the bone marrow or comet morphology in the liver. These results were formalized in the final study report received in May 2022, and, as a result of the study’s findings, we are not required to refund Orion any of the upfront fee.
Oaktree Credit Agreement
On May 11, 2021 (Closing Date), we entered into a Credit Agreement and Guaranty (as amended by that certain letter agreement on May 17, 2021, the Credit Agreement) with Oaktree Fund Administration, LLC as administrative agent (Oaktree) and the lenders party thereto (collectively, the Lenders) that provides for a five-year senior secured term loan facility in an aggregate original principal amount of up to $125.0 million, available to us in five tranches (collectively, the Term Loans). Upon entering into the Credit Agreement in May 2021, we borrowed $15.0 million in term loans from the Lenders (Tranche A-1 Term Loan), upon receipt of written acceptance by the FDA of our NDA filing relating to the use of ganaxolone in CDD in September 2021, we borrowed $30.0 million of tranche A-2 term loans from the Lenders (Tranche A-2 Term Loan), and in March 2022, we borrowed $30.0 million in term loans from the Lenders that became available as a result of the approval by the FDA of ZTALMY oral suspension for the treatment of seizures associated with CDD in patients two years of age and older (Tranche B Term Loan). Under the terms of the Credit Agreement, we may, at our sole discretion, borrow from the Lenders up to an additional $50.0 million in term loans subject to certain milestone events, as follows:
| ● | Through June 30, 2023, $25.0 million of tranche C term loans subject to the completion of one or more financings, including through the issuance of common stock, convertible debt, subordinated debt, a synthetic royalty or a sublicense in which we receive gross proceeds in an aggregate amount of at least $40.0 million and net proceeds in an aggregate amount of at least $36.0 million. In addition, the availability of this tranche is subject to either our current Phase 3 trial in RSE or a Phase 3 trial in TSC achieving statistical significance (p value < 0.05) across all primary endpoints and ganaxolone must be generally well tolerated, with a safety profile generally consistent with previous clinical trials. |
| ● | Through December 31, 2023, $25.0 million of tranche D term loans subject to us earning an aggregate of at least $50 million in net product revenue in the United States for a trailing six consecutive months. |
The Term Loans will bear interest at a fixed per annum rate (subject to increase during an event of default) of 11.50% and are scheduled to mature on the fifth anniversary of the Closing Date (Maturity Date). In addition, at the time of funding of any tranche of the Term Loans, we are required to pay an upfront fee of 2.0% of the aggregate principal amount being funded. We are required to make quarterly interest payments until the Maturity Date. We are also required to make principal payments, which are payable in quarterly installments beginning on the last day of the first quarter ending after the third anniversary of the Closing Date in an amount equal to 5.0% of the aggregate amount of the Term Loans outstanding on the date of the first such quarterly principal payment and continuing until the Maturity Date, on which date all outstanding Term Loans and other amounts owed under the Credit Agreement will be required to be paid in full. A commitment fee of 75 basis points per annum began to accrue on each of the tranche B, C and D commitments for the period beginning 120 days after the funding date of the tranche A-2 term loans and continues to accrue until the applicable tranche is either funded or terminated. The funding date of the tranche A-2 term loans was September 27, 2021, and as such, we began accruing the commitment fees for tranche B, C, and D term loans on January 25, 2022. We drew down the additional $30.0 million of tranche B term loans in March 2022, and paid less than $0.1 million in commitment fees related to tranche B term loans. At March 31, 2022, we have less than $0.1 million accrued for commitment fees related to tranche C and D term loans.
34
BARDA Contract
In September 2020, we and BARDA entered into the BARDA Contract, under which we received an award of up to an estimated $51 million for development of IV-administered ganaxolone for the treatment of RSE. The BARDA Contract provides for funding to support, on a cost-sharing basis, the completion of a Phase 3 clinical trial of IV-administered ganaxolone in patients with RSE, which covers the RAISE trial, funding of pre-clinical studies to evaluate IV-administered ganaxolone as an effective treatment for RSE due to chemical nerve gas agent exposure, and funding of certain ganaxolone manufacturing scale-up and regulatory activities. In March 2022, we entered into an amendment with BARDA to extend the end date of our base performance period for funding under the BARDA Contract from September 1, 2022 to December 31, 2023.
The BARDA Contract consists of an approximately two-year base period, and an extension period through December 31, 2023 per the amendment described above, during which BARDA will provide up to approximately $21 million of funding for the RAISE trial on a cost share basis and funding of additional preclinical studies of ganaxolone in nerve agent exposure models. Following successful completion of the RAISE trial and preclinical studies in the base period and extension period, the BARDA Contract provides for approximately $30 million of additional BARDA funding for three options in support of ganaxolone manufacturing, supply chain, clinical, regulatory and toxicology activities. Under the BARDA Contract, we will be responsible for cost sharing in the amount of approximately $33 million and BARDA will be responsible for approximately $51 million if all development options are completed. The contract period-of-performance (base period plus option exercises) is up to approximately five years.
Equity Distribution Agreement
In October 2017, we entered into an Equity Distribution Agreement (Prior EDA) with JMP Securities LLC (JMP), under which JMP, as our exclusive agent, at our discretion and at such times that we may determine from time to time, may sell over a three-year period from the execution of the agreement up to a maximum of $50 million of shares of our common stock. On July 9, 2020, we entered into a new Equity Distribution Agreement (New EDA) with JMP to create an at the market equity program under which we from time to time may offer and sell shares of our common stock having an aggregate offering price of up to $60 million through or to JMP. Subject to the terms and conditions of the New EDA, JMP will use its commercially reasonable efforts to sell shares of our common stock from time to time, based upon our instructions. JMP will be entitled to a commission of up to 3.0% of the gross proceeds from each sale of shares of our common stock. The New EDA superseded and terminated the Prior EDA effective immediately upon effectiveness of our shelf registration statement on Form S-3 (File No. 333-239780) filed with the SEC on July 9, 2020 and declared effective by the SEC on July 27, 2020. We did not sell any shares of our common stock during the three months ended March 31, 2022 or during the year ended December 31, 2021 under the New EDA.
IP License Agreement
In March 2022, we entered into an exclusive patent license agreement (License Agreement) with Ovid Therapeutics Inc. (Ovid). Under the License Agreement, we have an exclusive, non-transferable (except as provided in the License Agreement), royalty-bearing, sublicensable license under certain of Ovid’s patent(s) and patent applications to develop, make, have made, commercialize, promote, distribute, sell, offer for sale and import, ganaxolone, including any analogues or derivatives, including its salts, and pharmaceutical formulations of the foregoing (Licensed Products), in the U.S., the member states of the EU, Iceland, Lichtenstein, Norway, the United Kingdom, and Switzerland (Territory) for the treatment of CDD in humans (Field). Under the License Agreement, we have the sole right and responsibility for, and control over, all development, manufacturing, and commercialization activities, including all regulatory activities, with respect to the Licensed Products in the Field in the Territory. In addition, all regulatory approvals and related filings with respect to the Licensed Products in the Field in the Territory will be in the name of, and be owned solely by, us. We were required, at Ovid’s option exercisable in accordance with the License Agreement, to (i) pay to Ovid the sum of $1.5 million in cash; or (ii) issue to Ovid 123,255 shares of our common stock, which option to obtain shares of our common stock was exercisable within the five-business day period following the filing of our Annual Report on Form 10-K for the year ended December 31, 2021 on March 24, 2022. On March 29, 2022, we issued 123,255 shares of our common stock to Ovid, per Ovid’s option in accordance with the License Agreement. As
35
such, we recorded $1.2 million of IP license fee expenses related to the Ovid License Agreement in the three months ended March 31, 2022.
The License Agreement also provides for payment of royalties by us to Ovid in the low single digits on net sales by us, our affiliates and sublicensees, of Licensed Products in the Field in the Territory. Such royalties are subject to reduction in the event of generic competition in accordance with the License Agreement. We may terminate the License Agreement at any time without cause on thirty days’ prior written notice. Either party may terminate the License Agreement for the other party’s material breach or insolvency subject to certain cure periods. Also, Ovid has the right to terminate the License Agreement if there has not been a first commercial sale of any Licensed Products in the Field in the Territory on or before June 30, 2025. In the event of termination, all licenses granted under the License Agreement will terminate.
Cash Flows
Operating Activities. Cash used in operating activities increased to $27.7 million for the three months ended March 31, 2022 compared to $16.2 million for the three months ended March 31, 2021. Excluding the noncash impacts primarily related to depreciation and amortization, debt issuance costs, stock-based compensation, cost of license agreement and changes in the net contract assets/liabilities related to the Orion Collaboration Agreement, the change in cash used in operating activities for the three months ended March 31, 2022 compared to the same period in 2021, was primarily the result of decreases in the changes in accounts payable, accrued expenses and other long term-liabilities and an increase in operating expenses.
Investing Activities. Cash used by investing activities for the three months ended March 31, 2022 represents $0.1 million in purchases of property and equipment. Cash provided by investing activities for the three months ended March 31, 2021 represents the maturity of short-term investments of $1.5 million, partially offset by $0.4 million in deposits on property and equipment.
Financing Activities. Cash provided by financing activities for the three months ended March 31, 2022 includes $29.4 million in proceeds from notes payable, net of issuance costs, and $1.7 million in proceeds from the exercise of stock options. Cash provided by financing activities for the three months ended March 31, 2021 includes $0.2 million in proceeds from the exercise of stock options, partially offset by $0.1 million of debt issuance costs.
Funding Requirements
We have not achieved profitability since our inception, and we expect to continue to incur net losses for the foreseeable future. We expect our cash expenditures to increase in the near term as we fund the commercialization of ZTALMY and our continuing and planned clinical trials for ganaxolone.
We had cash and cash equivalents of $126.3 at March 31, 2022. We believe that our existing cash and cash equivalents on hand as of March 31, 2022 will be sufficient to fund our operating expenses, capital expenditure requirements and maintain the minimum cash balance required under our debt facility into the first quarter of 2023. However, we will need to secure additional funding in the future, from one or more equity or debt financings, government funding, collaborations, licensing transactions, other commercial transactions or other sources, such as the monetization of our Priority Review Voucher, in order to carry out all of our planned research and development activities with respect to ganaxolone. In order to meet these additional cash requirements, we may seek to sell additional equity or convertible debt securities that may result in dilution to our stockholders, or engage in federal contracts or other partnerships. If we raise additional funds through the issuance of convertible debt securities, these securities could have rights senior to those of our common stock and could contain covenants that restrict our operations. There can be no assurance that we will be able to obtain additional equity or debt financing on terms acceptable to us, if at all. Further, the continued spread of COVID-19 has also led to severe disruption and volatility in the global capital markets, which could increase our cost of capital and adversely affect our ability to access the capital markets in the future. Our failure to obtain sufficient funds on acceptable terms when needed could have a negative impact on our business, results of operations, and financial condition.
36
Our future capital requirements will depend on many factors, including:
| ● | the timing and success of our commercialization of ZTALMY; |
| ● | the ongoing and planned development, formulation and commercialization activities related to ganaxolone; |
| ● | the effects of the COVID-19 pandemic on our business, the medical community and the global economy; |
| ● | the scope, progress, results and costs of researching and developing ganaxolone or any other future product candidates, and conducting preclinical studies and clinical trials; |
| ● | the timing of, and the costs involved in, obtaining regulatory approvals for ganaxolone or any other future product candidates; |
| ● | the cost of commercialization activities for ZTALMY or any other future product candidates that are approved for sale, including marketing, sales and distribution costs; |
| ● | the cost of manufacturing and formulating ganaxolone, or any other future product candidates, to internal and regulatory standards for use in preclinical studies, clinical trials and, if approved, commercial sale; |
| ● | our ability to establish and maintain strategic collaborations, licensing or other arrangements and the financial terms of such agreements; |
| ● | our ability to receive funding under the BARDA Contract; |
| ● | our expectations regarding the amount and timing of milestone and royalty payments pursuant to our exclusive license agreement with Orion for the commercialization of ganaxolone in Europe; |
| ● | our eligibility for additional debt tranches under the Credit Agreement with Oaktree; |
| ● | any product liability, infringement or other lawsuits related to our product candidates and, if approved, products; |
| ● | capital needed to attract and retain skilled personnel; |
| ● | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims, including litigation costs and the outcome of such litigation; and |
| ● | the timing, receipt and amount of sales of, or royalties on, approved products, including payments payable to Ovid. |
Please see the Risk Factors section included in our Annual Report on Form 10-K for the year ended December 31, 2021 filed with the SEC on March 24, 2022 for additional risks associated with our substantial capital requirements.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that are material to investors.
37
Discussion of Critical Accounting Policies and Significant Judgments and Estimates
The preparation of financial statements in conformity with GAAP requires us to use judgment in making certain estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities and the reported amounts of revenues and expenses in our financial statements and accompanying notes. Critical accounting policies are those that are most important to the portrayal of our financial condition and results of operations and require difficult, subjective and complex judgments by management in order to make estimates about the effect of matters that are inherently uncertain. During the three months ended March 31, 2022, there were no significant changes to our critical accounting policies from those described in our annual financial statements for the year ended December 31, 2021, which we included in our Annual Report on Form 10-K and was filed with the SEC on March 24, 2022.
Item 3. Quantitative and Qualitative Disclosures About Market Risk
We are a smaller reporting company as defined by Rule 12b-2 of the Securities Exchange Act of 1934, as amended (Exchange Act) and are not required to provide the information under this item.
Item 4. Controls and Procedures
(a) Evaluation of Disclosure Controls and Procedures.
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures (as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of the end of the period covered by this Quarterly Report on Form 10-Q to ensure that the information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms, and that information required to be disclosed in the reports we file or submit under the Exchange Act is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, to allow timely decisions regarding required disclosures. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost benefit relationship of possible controls and procedures. Based on such evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that our disclosure controls and procedures were effective at the reasonable assurance level as of March 31, 2022.
(b) Changes in Internal Control Over Financial Reporting
There was no change in our internal control over financial reporting identified in connection with the evaluation required by Rule 13a-15(d) and 15d-15(d) of the Exchange Act that occurred during the quarter ended March 31, 2022 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
38
PART II
OTHER INFORMATION
Item 1. Legal Proceedings
From time to time, we may become subject to litigation and claims arising in the ordinary course of business. We are not currently a party to any material legal proceedings, and we are not aware of any pending or threatened legal proceedings against us that we believe could have a material adverse effect on our business, operating results or financial condition.
Item 1A. Risk Factors
Except as set forth below, there have been no material changes to the risk factors previously disclosed in our Annual Report on Form 10-K for the fiscal year ended December 31, 2021.
We have multiple ganaxolone drug products in development, and until such products are approved by regulatory authorities, there remains the risk that the drug product quality requirements may not support continued clinical investigation and result in delays or termination of such clinical studies, and product approvals.
We currently have multiple ganaxolone drug products in clinical development, including an oral suspension, IV solution and a new formulation that is being utilized in an ongoing Phase 1 trial with top-line data expected to be announced in mid-2022. While we strive to develop a full understanding of manufacturing processes used, as well as the resultant product quality attributes, there is a risk that problems may arise over the course of development which could render a given drug product non-viable. Such problems could relate to manufacturing reproducibility, scale-up challenges, drug product chemical or physical stability issues. Related quality requirements may not support continued clinical investigation and result in delays or termination of such clinical studies and product approvals. Such quality requirements can include physical and chemical attributes of the drug product, stability and shelf life, microbial and other contamination, including adverse impact of drug product packaging and administration devices. These problems could result in unacceptable manufacturing economics, or direct concerns related to drug product safety or efficacy. For example, we announced in February 2022 a product supply interruption for our IV ganaxolone clinical supplies. Routine monitoring of stability batches of IV clinical supply material showed visible particulates of aluminum phosphate in the drug solution, which led to a pause in recruitment for the RAISE trial. In May 2022, we announced that the RAISE trial has resumed utilizing new batches of the current IV formulation of ganaxolone. In connection with the resumption of the trial and in consultation with the FDA, Marinus has implemented a 12- month shelf life; refrigerated storage conditions for the entire duration of clinical use, including storage at the clinical sites; and frequent testing for visible particles. If we experience issues with product quality requirements and we are unable to resolve these issues in a timely manner or at all, we may need to delay or terminate our RAISE or other clinical trials with the current IV formulation, which could further delay our clinical development plans and future product approvals.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds
On March 29, 2022, pursuant to an exclusive patent license agreement with Ovid Therapeutics Inc. (Ovid), we issued 123,255 shares of our common stock to Ovid. The issuance of the shares was exempt from the registration requirements of the Securities Act of 1933, as amended (the Securities Act), and the shares of common stock issued to Ovid were offered and sold without registration under the Securities Act pursuant to the exemption provided by Section 4(a)(2) of the Securities Act and Rule 506 promulgated thereunder as transactions not involving a public offering, as well as similar exemptions under applicable state securities laws, in reliance upon the following facts: no general solicitation was used in the offer or sale of such securities; the recipient of the securities had adequate access to information about us; the recipient of such securities represented its acquisition thereof as principal for its own account and its lack of any arrangements or understandings regarding the distribution of such securities; the recipient of such securities represented its capability of evaluating the merits of an investment in our securities due to its knowledge, sophistication and experience in business and financial matters; and such securities were issued as restricted securities with restricted legend referring to the Securities Act.
39
Item 3. Defaults upon Senior Securities
None.
Item 4. Mine Safety Disclosures
Not applicable.
Item 5. Other Information
Not applicable.
40
Item 6. Exhibits
Exhibit |
| Exhibit Description |
3.1 | ||
3.2 | ||
3.3 | ||
3.4 | ||
3.5 | ||
3.6 | ||
3.7 | ||
3.8 | ||
4.1 | ||
10.1* | ||
10.2 | ||
10.3+ | ||
31.1 | ||
31.2 | ||
32.1 | ||
101.INS | XBRL Instance Document – the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document | |
101.SCH | XBRL Taxonomy Extension Schema | |
101.CAL | XBRL Taxonomy Extension Calculation Linkbase | |
101.DEF | XBRL Taxonomy Extension Definition Linkbase | |
101.LAB | XBRL Taxonomy Extension Labels Linkbase | |
101.PRE | XBRL Taxonomy Extension Presentation Linkbase | |
104 | Cover Page Interactive Data File formatted as Inline XBRL and contained in Exhibit 101 |
* Portions of this exhibit have been omitted in compliance with Item 601 of Regulation S-K.
+ Indicates management contract or compensatory plan.
41
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
Signature |
| Title |
| Date |
/s/ SCOTT BRAUNSTEIN, M.D. | President and Chief Executive Officer (principal executive officer) and Director | May 12, 2022 | ||
Scott Braunstein, M.D. | ||||
/s/ STEVEN PFANSTIEL | Chief Financial Officer and Treasurer (principal financial and accounting officer) | May 12, 2022 | ||
Steven Pfanstiel |
42
Similar companies
See also JOHNSON & JOHNSON - Annual report 2023 (10-K 2023-01-01) Annual report 2023 (10-Q 2023-10-01)See also ELI LILLY & Co - Annual report 2024 (10-K 2024-12-31) Annual report 2023 (10-Q 2023-09-30)
See also PFIZER INC - Annual report 2022 (10-K 2022-12-31) Annual report 2023 (10-Q 2023-10-01)
See also AbbVie Inc. - Annual report 2022 (10-K 2022-12-31) Annual report 2023 (10-Q 2023-09-30)
See also NOVO NORDISK A S