Lineage Cell Therapeutics, Inc. - Annual Report: 2020 (Form 10-K)
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
☒ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d)
OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2020
OR
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d)
OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from___________ to __________
Commission file number 001-12830
Lineage Cell Therapeutics, Inc.
(Exact name of registrant as specified in its charter)
| California | 94-3127919 | |
| (State
or other jurisdiction of incorporation or organization) |
(I.R.S.
Employer Identification No.) |
2173 Salk Avenue, Suite 200
Carlsbad, California 92008
(Address of principal executive offices) (Zip Code)
Registrant’s telephone number, including area code (442) 287-8990
Securities registered pursuant to Section 12(b) of the Act
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
| Common stock | LCTX | NYSE American |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☐ | Accelerated filer ☐ |
| Non-accelerated filer ☒ | Smaller reporting company ☒ |
| Emerging growth company ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act): Yes ☐ No ☒
As of June 30, 2020, the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market value of the registrant’s voting and non-voting common equity held by non-affiliates of the registrant was approximately $93.9 million.
The number of common shares outstanding as of March 5, 2021 was .
Lineage Cell Therapeutics, Inc.
Table of Contents
PART I
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K (this “Report”) contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), that involve substantial risks and uncertainties. The forward-looking statements are contained principally in Part I, Item 1. “Business,” Part I, Item 1A. “Risk Factors,” and Part II, Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” but are also contained elsewhere in this Report. In some cases, you can identify forward-looking statements by the words “may,” “might,” “will,” “could,” “would,” “should,” “expect,” “intend,” “plan,” “objective,” “anticipate,” “believe,” “estimate,” “predict,” “project,” “potential,” “continue” and “ongoing,” or the negative of these terms, or other comparable terminology intended to identify statements about the future. These statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements. Although we believe that we have a reasonable basis for each forward-looking statement contained in this Report, we caution you that these statements are based on a combination of facts and factors currently known by us and our expectations of the future, about which we cannot be certain. Forward-looking statements include statements about:
| ● | our plans to research, develop and commercialize our product candidates; | |
| ● | the initiation, progress, success, cost and timing of our clinical trials and product development activities; | |
| ● | the therapeutic potential of our product candidates, and the disease indications for which we intend to develop our product candidates; | |
| ● | our ability and timing to advance our product candidates into, and to successfully initiate, conduct, enroll and complete, clinical trials; | |
| ● | our ability to manufacture our product candidates for clinical development and, if approved, for commercialization, and the timing and costs of such manufacture; | |
| ● | the performance of third parties in connection with the development and manufacture of our product candidates, including third parties conducting our clinical trials as well as third-party suppliers and manufacturers; | |
| ● | the potential of our cell therapy platform, and our plans to apply our platform to research, develop and commercialize our product candidates; | |
| ● | our ability to obtain funding for our operations, including funding necessary to initiate and complete clinical trials of our product candidates; | |
| ● | the size and growth of the potential markets for our product candidates and our ability to serve those markets; | |
| ● | the potential scope and value of our intellectual property rights; | |
| ● | our ability, and the ability of our licensors, to obtain, maintain, defend and enforce intellectual property rights protecting our product candidates, and our ability to develop and commercialize our product candidates without infringing the proprietary rights of third parties; | |
| ● | our ability to recruit and retain key personnel; | |
| ● | the effects of the COVID-19 pandemic on our operations; and | |
| ● | other risks and uncertainties, including those described under Part I, Item 1A. Risk Factors of this Report. |
| 1 |
You should refer to “Item 1A. Risk Factors” in this Report for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all. The forward-looking statements in this Report represent our views as of the date of this Report. We anticipate that subsequent events and developments may cause our views to change. However, while we may elect to update these forward-looking statements at some point in the future, we undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law. You should, therefore, not rely on these forward-looking statements as representing our views as of any date subsequent to the date of this Report.
You should read this Report and the documents that we reference in this Report completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
This Report also contains market data, industry forecasts and other data made by independent parties and by us relating to market size and growth and other data about our industry. This data involves a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. In addition, projections, assumptions and estimates of our future performance and the future performance of the markets in which we operate are necessarily subject to a high degree of uncertainty and risk.
All brand names or trademarks appearing in this Report are the property of their respective owners. Solely for convenience, the trademarks and trade names in this Report are referred to without the symbols ® and TM, but such references should not be construed as any indication that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto.
Unless the context requires otherwise, references in this report to “Lineage,” “we,” “us,” and “our” refer to Lineage Cell Therapeutics, Inc. and its consolidated subsidiaries.
RISK FACTOR SUMMARY
Below is a summary of the material factors that make an investment in our stock speculative or risky. This summary does not address all of the risks that we face. Additional discussion of the risks summarized in this risk factor summary, and other risks that we face, can be found below under the heading “Risk Factors” in Item 1A of Part I of this Report and should be carefully considered, together with other information in this Report and our other filings with the Securities and Exchange Commission before making investment decisions regarding our common shares.
| ● | We have incurred operating losses since inception, and we do not know if or when we will attain profitability. | |
| ● | We will continue to spend a substantial amount of our capital on research and development, but we might not succeed in developing products and technologies that are useful in medicine. | |
| ● | The amount and pace of research and development work that we can do or sponsor, and our ability to commence and complete clinical trials required to obtain regulatory approval to market our therapeutic and medical device products, depends upon the amount of funds we have. | |
| ● | We will need to issue additional equity or debt securities in order to raise additional capital needed to pay our operating expenses. | |
| ● | We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, including anti-kickback and false claims laws, transparency laws, and health information privacy and security laws. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties. | |
| ● | If we do not receive regulatory approvals, we will not be permitted to sell our therapeutic and medical device products. |
| 2 |
| ● | Government-imposed bans or restrictions and religious, moral, and ethical concerns about the use of hES cells could prevent us from developing and successfully marketing stem cell products. | |
| ● | We expect that the commercial opportunity for some of our products may depend on our ability to obtain reimbursement and continued coverage from various payors, including government entities and insurance companies. | |
| ● | Clinical studies are costly, time consuming and are subject to risks that could delay or prevent commercialization of our current or future product candidates. | |
| ● | Clinical and preclinical drug development involves a lengthy and expensive process with an uncertain outcome. The results of early preclinical trials and clinical trials of our product candidates are not necessarily predictive of future results. Our product candidates may not have favorable results in later clinical trials, if any, or receive regulatory approval on a timely basis, if at all. | |
| ● | Interim, topline and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data. | |
| ● | Our intellectual property may be insufficient to protect our products. | |
| ● | If we are unable to obtain and enforce patents and to protect our trade secrets, others could use our technology to compete with us, which could limit opportunities for us to generate revenues by licensing our technology and selling products. | |
| ● | We may become dependent on possible future collaborations to develop and commercialize many of our product candidates and to provide the regulatory compliance, sales, marketing and distribution capabilities required for the success of our business. | |
| ● | Because we are engaged in the development of pharmaceutical and stem cell therapy products, the price of our common shares may rise and fall rapidly. | |
| ● | Current economic and stock market conditions may adversely affect the price of our common shares. |
| ITEM 1. | BUSINESS |
Overview
We are a clinical-stage biotechnology company developing novel cell therapies for unmet medical needs. Our focus is to develop therapies for degenerative retinal diseases, neurological conditions associated with demyelination, and aiding the body in detecting and combating cancer. Specifically, Lineage is testing therapies to treat dry age-related macular degeneration, spinal cord injuries, and non-small cell lung cancer. Our programs are based on our proprietary cell-based therapy platform and associated development and manufacturing capabilities. From this platform, we develop and manufacture specialized, terminally or functionally differentiated human cells from established and well-characterized pluripotent cell lines. These differentiated cells are transplanted into a patient either to replace or support cells that are dysfunctional or absent due to degenerative disease or traumatic injury, or are administered as a means of helping the body mount an effective immune response to cancer.
Product Candidates & Other Programs
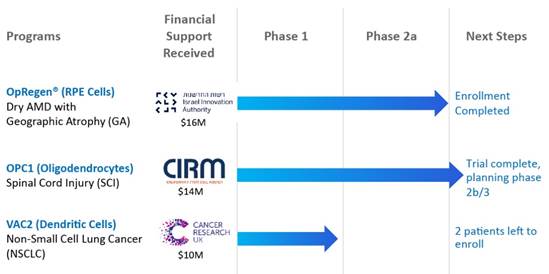
We have three allogeneic, or “off-the-shelf,” cell therapy programs in clinical development:
| ● | OpRegen®, a retinal pigment epithelium (“RPE”) cell replacement therapy currently in a Phase 1/2a multicenter clinical trial for the treatment of advanced dry age-related macular degeneration (“AMD”) with geographic atrophy (“GA”). There currently are no therapies approved by the U.S. Food and Drug Administration (“FDA”) for dry AMD, which accounts for approximately 85-90% of all AMD cases and is the leading cause of blindness in people over the age of 60. |
| 3 |
| ● | OPC1, an oligodendrocyte progenitor cell therapy currently in the long-term follow-up portion of a Phase 1/2a multicenter clinical trial for acute spinal cord injuries (“SCI”). This clinical trial has been partially funded by the California Institute for Regenerative Medicine. | |
| ● | VAC2, an allogeneic cancer immunotherapy of antigen-presenting dendritic cells currently in a Phase 1 clinical trial in non-small cell lung cancer. This clinical trial is being funded and conducted by Cancer Research UK, the world’s largest independent cancer research charity. |
In addition to seeking to create value for shareholders by developing product candidates and other technologies through our clinical development programs, we also seek to create value from our technologies through partnering and strategic transactions. We founded two companies that later became publicly traded companies: OncoCyte Corporation (“OncoCyte”) and AgeX Therapeutics, Inc. (“AgeX”).
During the year ended December 31, 2020, we received approximately $12.6 million in gross proceeds in connection with our sale of shares of OncoCyte and AgeX. In August 2020, we also received $24.6 million from Juvenescence Limited (“Juvenescence”), representing principal and accrued interest under a promissory note we received in connection with our sale of AgeX shares to Juvenescence in August 2018.
We no longer hold any common stock in AgeX. The value of our OncoCyte holdings as of March 5, 2021, was approximately $4.2 million, based on the closing price of its common stock on that date. In this Report, see Part I, Item 1A, “Risk Factors—Risks Related to Our Business Operations and Capital Requirements—The value of our investments in public companies fluctuates based on their respective stock prices and could be negatively affected by poor business performance.”
Though our principal focus is on advancing our three cell therapy programs currently in clinical development, we may seek to create additional value through corporate transactions, as we have in the past, or by initiating new programs using our protocols or with new protocols and cell lines.
Corporate Information
Lineage is incorporated in the State of California. Our common shares trade on the NYSE American and the Tel Aviv Stock Exchange under the symbol “LCTX.” Our principal executive offices are at 2173 Salk Avenue, Suite 200, Carlsbad, CA 92008, and our phone number at that address is (442) 287-8990. Our website address is www.lineagecell.com. The information on, or that can be accessed through our website is not part of this Report. Lineage routinely uses its website as a means of disclosing material non-public information and for complying with its disclosure obligations under Regulation FD. We also make available, free of charge through our website, our most recent annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and any amendments to those reports as soon as reasonably practicable after the reports are electronically filed with or furnished to the Securities and Exchange Commission.
2020 Highlights
We achieved numerous strategic accomplishments during 2020, including advancing clinical trials and product development in several key programs.
| ● | In May 2020, we announced the early exercise of our option with Cancer Research UK to bring the VAC immuno-oncology platform in-house. | |
| ● | In June 2020, we announced the first known finding of retinal tissue restoration in a patient who received an RPE cell transplant. | |
| ● | In October 2020, we reported encouraging preliminary Phase 1 clinical study results with VAC2 for the treatment of non-small cell lung cancer with high levels of antigen-specific immunogenicity observed. | |
| ● | In November 2020, we completed enrollment in a 24 patient Phase 1/2a clinical study of OpRegen for the treatment of dry AMD with GA with encouraging preliminary signs of tolerability and efficacy. | |
| ● | In December 2020, we announced that we had been able to make significant manufacturing improvements to our OPC1 acute SCI program, including better controlled processes, enhanced purity, potency, and scale, and to the development of a “ready-to-inject” formulation, substantially decreasing logistical burden at the point of care and enabling use at a much larger number of treatment centers. |
| 4 |
Business Strategy
Our goal is to become a leading cell therapy company by developing allogeneic, or “off-the-shelf,” treatments that are comprised of differentiated cells derived from pluripotent cell lines, which have been directed to become specific cell types and use those cells as treatments to restore diseased or diminished functions, such as impaired vision, loss of movement and sensation, or to increase immune response to tumors. Significant near-term activities that underlie our business strategy include:
| ● | Presenting new and accumulated OpRegen data from the ongoing Phase 1/2a clinical study on two occasions during the first and second quarters of 2021; | |
| ● | Completing VAC2 patient enrollment in the ongoing Phase 1 clinical study for the treatment of non-small cell lung cancer by the end of the first half of 2021; | |
| ● | Evaluating delivery improvements for our OPC1 program, which combined with our “ready-to-inject” formulation, will enable access to a greater number of clinical sites, currently ongoing and throughout 2021; | |
| ● | Meeting with the FDA to discuss further development of the OPC1 program, including a late-stage clinical study, during the second half of 2021; | |
| ● | Evaluating opportunities for new VAC product candidates based on manufacturing improvements and product improvements, including newly discovered tumor antigens/neoantigens, throughout 2021; and | |
| ● | Evaluating partnership opportunities and expansion of existing external collaborations and identification of new collaborations for OpRegen, OPC1 and VAC2, currently ongoing and throughout 2021. |
Cell Therapy Technology
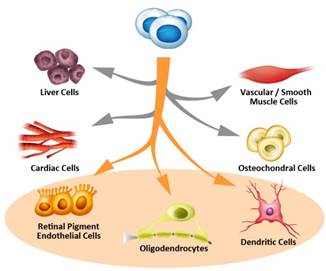
We believe we are a leader in pluripotent, cell-based asset development based on directed lineage derivation protocols and whole cell manufacturing capabilities. Pluripotent cells, which are widely published as capable of becoming any human cell type, have potential applications in many areas of medicine with large unmet patient needs, including certain age-related degenerative diseases and degenerative conditions for which there presently are no cures. We currently are focused on developing pluripotent cells into RPE cells, oligodendrocyte progenitor cells and dendritic cells.
Pluripotent Stem Cells
| 5 |
Unlike pharmaceuticals that require a narrowly defined molecular target, cellular therapies are often aimed at regenerating or replacing the entire affected cell or tissue and therefore, may have broader or more suitable applicability than many traditional pharmaceutical products. Small molecules and biologic therapies that require systemic delivery into the body often have unexpected results, or side effects, that can limit their usefulness. When cell replacement is locally administered, particularly to anatomical compartments, systemic side effects are usually not the primary concern. The risk profile of cell therapy more closely resembles that of transplant medicine, focused more on whether the transplanted cells are rejected by the body and whether the cells function as expected. We currently are using our pluripotent stem cells as starting material from which we derive three separate and specific cell types, each of which are product candidates currently in clinical testing.
We maintain an innovative cell therapy manufacturing facility in the Bio Park on the campus of the Hadassah University Hospital in Jerusalem, Israel. The facility includes process development laboratories and a state-of-the-art, cGMP manufacturing facility. It is designed and equipped to enable simultaneous cGMP processes and to produce a range of cell therapy products for human use in clinical trials as well as developing scale suitable for commercial launch. All cGMP manufacturing processes, including cell banks and product manufacturing for our cell therapy product candidates, are conducted in this facility.
Cell Therapy Product Candidates
OpRegen
OpRegen is our lead ophthalmic product candidate (currently in a Phase 1/2a clinical trial) for the treatment of advanced dry AMD with GA. AMD is a gradual, progressive, deterioration of the macula, the small sensitive area in the center of the retina that provides clear, high definition central vision. AMD affects over 30 million people worldwide and approximately 1.6 million people are diagnosed annually in the United States. It is a leading cause of vision loss in people over the age of 65 in the developed world. As the area of atrophy begins to include the fovea (the center of the macula), patients lose their central vision, making facial recognition, reading and driving difficult or impossible, and often resulting in legal blindness. The exact cause of dry AMD is unknown, but is thought to result from multiple factors, such as genetics, age and environmental effects. There are two clinical presentations of AMD, the dry form and the wet form, or neovascular form (growth of abnormal new blood vessels). Dry AMD typically advances slowly toward GA in which RPE cells and photoreceptors deteriorate over time. RPE cells support and nourish the retina by metabolizing waste by-products and producing a number of components useful for photoreceptor health and function. If the metabolic waste products accumulate, lesions known as drusen are generated. Approximately 85-90% of AMD patients suffer from dry AMD, for which there is no FDA-approved medical therapies. Dry AMD may also lead to wet AMD, a condition for which there are several FDA-approved treatments administered locally to inhibit the growth of new blood vessels, but these treatments are not effective nor approved for the treatment of dry AMD. Physicians often recommend a healthy diet, exercise and/or nutritional supplements for dry AMD, but nutritional supplements have shown limited efficacy in delaying the onset of more progressive disease in longer-term studies. The schematics below show a representation of the process of drusen formation and the goal of cell replacement therapy.
| 6 |
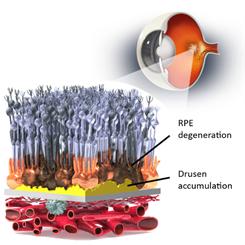
Dry AMD involves the loss of retina cells, creating an area of geographic atrophy (GA), which causes impaired vision and blindness
We believe one of the most promising approaches to treat dry AMD is to replace the layer of damaged RPE cells with new, healthy and functional RPE cells manufactured from a well-characterized cell line. OpRegen is a cell replacement therapy derived from our pluripotent cell technology in which our proprietary directed-differentiation methods convert pluripotent stem cells into nearly pure populations of RPE cells. Using this method, OpRegen is grown free of any animal products and consists of human RPE cells with high yield and purity that can be transplanted directly into the patient’s eye, where the patient’s own RPE cells are missing or dysfunctional. The OpRegen therapeutic approach is designed to replace damaged or lost RPE cells with the goal of slowing disease progression to preserve and/or restore visual function.
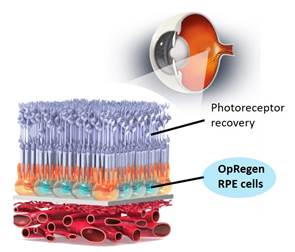
OpRegen is an injection of RPE cells delivered to the retina, to replace lost retinal cells and preserve or restore vision
Preclinical studies in the Royal College of Surgeons (RCS) rat model have shown that following a single subretinal injection, OpRegen as a suspension of cells rapidly organized into their natural monolayer structure and survived until the end of the study, which we believe is critical to the potential success of OpRegen in humans. Additionally, rats receiving OpRegen had objective evidence of improved optomotor tracking, indicating functional visual improvement compared to control animals.
OpRegen is intended to be an allogeneic, or “off-the-shelf,” product provided to retinal surgeons in an “easy-to-use” form for transplantation. We believe OpRegen could have a lasting benefit from a single administration, or once every several years. This approach differs from other investigational drugs for Dry AMD and approved agents currently marketed for wet AMD, such as Ranibizumab (Lucentis®) and Aflibercept (Eylea®), that require multiple, frequent intravitreal injections into the eye.
| 7 |
The patients in our ongoing Phase 1/2a clinical trial are 50 years of age or older, whose dry AMD has advanced to the GA stage, with absence of additional concomitant ocular disorders. The eye in which the disease has progressed the most is treated, while their other eye serves as a measure of disease progression. Following injection, the patients are followed for 12 months at specified intervals to evaluate the safety and tolerability of OpRegen.
Following the initial 12-month period, patients are evaluated at longer intervals for up to an additional five years following administration. A secondary objective of the clinical trial is to examine the ability of transplanted OpRegen to engraft, survive, and modulate disease progression in the patients. In addition to thorough characterization of visual function, several vision tests are used to quantify stabilization or improvements in visual function. We also perform anatomical evaluation imaging to assess the restoration of the structure of the retina.
Interim data from the first 12 subjects in Cohorts 1-3 have been encouraging and suggest that OpRegen RPE cells are generally well-tolerated when administered by subretinal injection in these legally blind patients with large areas of GA that have encompassed the foveal area. The surgical procedures were generally well-tolerated, with spectral domain optical coherence tomography (SD-OCT) images showing absorption of the subretinal fluid in the bleb less than 48 hours after surgery and healing of the site of retinal penetration by the cannula within a few weeks. Initial findings using a variety of imaging modalities suggest presence of cells in the subretinal space, an observation consistent with, and supported by, the data from preclinical studies of OpRegen. Findings on clinical examination by different imaging modalities show potential improvements in retinal structure, which could precede visual functional improvements. Though it is not definitively known at this time whether these changes represent engraftment and survival of the transplanted cells, data from the preclinical animal studies suggest this is the most likely scenario.
Importantly, in this safety-focused aspect of the trial, no unexpected ocular adverse events have been observed and those events expected to occur based on the procedures involved in OpRegen administration, such as vitrectomy, have been predominately mild in severity. The majority of these subjects had pre-existing epiretinal membranes (ERMs) at the time of trial enrollment and in most cases, experienced new or worsening ERMs following the surgical procedure, which is believed to be partially attributable to the route of administration via pars plana vitrectomy (PPV) and retinotomy. The majority were mild to moderate in severity, though two patients with severe ERM were successfully treated via a routine surgical procedure. These subjects are being monitored during trial follow-up. One instance of retinal detachment occurred in a patient who was legally blind prior to treatment. The event was not assigned as related to treatment, procedure or to the combination. The patient continued for a period of time in the trial following successful surgical repair but has since withdrawn due to other unrelated health issues. The independent data safety monitoring board approved moving to Cohort 4 based on the safety data from the Cohorts 1-3. Cohort 4 incorporates an additional variety of objective and subjective assessments to look for signs of potential efficacy as well as potential anatomical changes indicative of OpRegen cell function following implantation.
As described above, many of the adverse events (AEs) observed in subretinal procedures are likely related to the delivery technique utilized during the surgery. As previously described, in January 2019, we announced an exclusive partnership with Orbit Biomedical (now Gyroscope Therapeutics, Ltd.) to assess its FDA-cleared Orbit Subretinal Delivery System (SDS), a single-use vitrectomy-free delivery device designed to deliver products to the subretinal space for the administration of OpRegen within the ongoing clinical trial. The device allows for access to the subretinal space via a sclerotomy and suprachoroidal approach, which means that there are no openings created into the vitreous chamber. This could eliminate the possibilities of new or worsening epiretinal membranes and exacerbation or generation of a cataract, both known issues with the older standard method of delivery. We believe that the use of this device could significantly decrease the number of adverse events and improve retention and dose control of OpRegen in our clinical trials.
We completed enrollment in Cohorts 1-3 (12 patients) in the middle of 2018 and as previously reported, OpRegen was well tolerated with no unexpected systemic serious adverse events (SAEs) or ocular AEs. Importantly, there were several patients that exhibited improved retinal structure, reduction in drusen, alterations in the pattern of GA progression and indications of long-term survival of the OpRegen cells. We began enrollment of Cohort 4 (targeted for an additional 12 patients) shortly thereafter and treated three patients via the traditional route of administration. In 2019, we amended our clinical protocol to incorporate the Orbit SDS and our new thaw and inject formulation into our Phase 1/2a clinical trial. In February 2020, we announced that after reviewing promising preliminary data from the ongoing OpRegen Phase 1/2a clinical trial, our independent data safety monitoring board removed the protocol-mandated treatment stagger. The COVID pandemic slowed the rate of patient accrual but study enrollment was completed on November 10, 2020, with the treatment of the twelfth Cohort 4 patient, seven via the Orbit SDS and five via PPV/retinotomy. Five different surgeons at four centers successfully delivered OpRegen using the Orbit SDS and there were no unexpected AEs. Encouraging structural and clinical changes in these better vision patients, including better visual acuity and increased reading speed, are being followed and updates will be provided at major medical meetings or as findings merit.
| 8 |
In June 2020, we were able to report the first known example of retinal restoration following OpRegen administration in a Cohort 4 patient who was treated via the PPV/retinotomy route, with the findings confirmed by several independent reviewers. It is hypothesized that photoreceptor cells in the transition areas at the boundary of the GA are dysfunctional and dying, but not completely lost. The addition of new RPE cells may restore the microenvironment in surrounding tissue and contribute to the possibility of restoring function to existing cells that otherwise, if left untreated, would inevitably progress to further expansion of the atrophic region. Specifically, in this patient, the area of GA assessed at nine months following OpRegen treatment was approximately 25% smaller than the patient’s pre-treatment baseline. As reported in November at the 2020 American Academy of Ophthalmology (AAO) Annual Meeting, this patient continues to show signs of a smaller area of GA and improved visual acuity. This unprecedented finding supports the view that dry AMD is not an irreversible, degenerative condition and that some portion of diseased retinal tissue may be recoverable in atrophic end-stage disease patients.
With enrollment complete, patients are being followed for safety and efficacy as per protocol. We plan to present OpRegen data to the FDA in the third quarter of 2021 for discussion about a subsequent, comparative clinical trial.
OPC1
OPC1 is our lead product candidate for the treatment of acute spinal cord injury (“SCI”). SCI occurs when the spinal cord is subjected to a severe crush or contusion injury, such as that caused by a car or motorcycle accident and typically results in severe functional impairment, including limb paralysis, aberrant pain signaling, and loss of bladder and sexual function. There are approximately 18,000 new spinal cord injuries annually in the U.S. (NSCIC SCI Facts and Figures at a Glance (2019)), and there are currently no FDA-approved drugs specifically for the treatment of SCI, although methylprednisolone, a corticosteroid generally used as an anti-inflammatory drug, is sometimes prescribed on an off-label basis to reduce acute inflammation in the injured spinal cord immediately after injury. It is believed that to effect substantial benefit in treating this complex injury, multiple mechanisms of action are required, such as introduction of biologics that preserve surviving neurons and stimulate new nerve axon outgrowth, suppression of lesion formation at the injury site, generation of new blood vessels to repair the ischemic damage from injury, and myelination of the demyelinated and newly formed nerve axons. A key therapeutic target in SCI is replacement of oligodendrocytes that are selectively lost at the injury site. As the sole source of the insulating protein myelin in the brain and spinal cord, oligodendrocytes wrap around nerve axons and allow conduction of electrical impulses throughout the central nervous system (“CNS”).
OPC1 is an oligodendrocyte progenitor cell therapy derived from our pluripotent cell technology under Current Good Manufacturing Practice (“cGMP”) conditions using a directed differentiation method. These cells are stored frozen until ready for use and prepared for direct administration into the injured spinal cord. Based on preclinical studies, when OPC1 is transplanted into the injured spinal cord, the cells undergo further maturation to generate a replacement population of oligodendrocytes at the injury site that are capable of remyelinating denuded and newly formed nerve axons. Prior to their maturation, the transplanted oligodendrocyte progenitor cells stimulate additional reparative processes, including promotion of neuron survival and nerve axon outgrowth, and induction of blood vessel formation in and around the injury site. In addition, OPC1 cells rapidly migrate from the injection point to the injury site where they generate a supportive tissue matrix and suppress cavitation. Cavitation is a destructive process that occurs within the spinal cord following SCI, and typically results in permanent loss of motor and sensory function. A patient with cavitation can develop a condition known as syringomyelia, which results in additional neurological and functional damage to the patient and can result in chronic pain. Based on the multiple reparative properties associated with OPC1, we believe this candidate cell therapy product is ideally suited to treat neurological conditions such as SCI and other demyelination and demyelination disorders of the CNS.
Under a grant for clinical development, the development of OPC1 has been supported by $14.3 million in funds from the California Institute for Regenerative Medicine (“CIRM”), from 2014 through the date of this Report. We intend to apply for additional grants from CIRM for the program’s continued development.
| 9 |
Prior to its acquisition, Asterias tested OPC1 in two clinical trials: a five patient Phase 1 safety trial and a 25-patient Phase 1/2a dose escalation trial, which we call the SCiStar trial. The SCiStar trial was an open-label, single-arm trial testing three sequential escalating doses of OPC1 administered at up to 20 million OPC1 cells with subacute, C-4 to C-7, motor complete (AIS-A or AIS-B) cervical SCI. These individuals have essentially lost all movement below their injury site and experience severe paralysis of the upper and lower limbs. AIS-A patients have lost all motor and sensory function below their injury site, while AIS-B patients have lost all motor function but may retain some minimal sensory function below their injury site. OPC1 was administered 21 to 42 days post-injury. Patients continue to be followed by neurological exams and imaging procedures to assess the safety and activity of the product. Enrollment was completed in December 2017 and consisted of five cohorts:
| Cohort | Injury Type; OPC1 Dose | # of Patients | ||
| Cohort 1 | AIS-A; 2 million OPC1 cells (low dose for safety evaluation) | 3 | ||
| Cohort 2 | AIS-A; 10 million OPC1 cells | 6 | ||
| Cohort 3 | AIS-A; 20 million OPC1 cells* | 6 | ||
| Cohort 4 | AIS-B; 10 million OPC1 cells | 6 | ||
| Cohort 5 | AIS-B; 20 million OPC1 cells* | 4 |
* One patient from Cohort 3 and one patient from Cohort 5 were administered 10 million cells.
In January 2019, top-line 12-month data from the SCiStar trial were announced by Asterias, which included the following key findings:
| ● | Positive Safety Profile. Magnetic resonance imaging (“MRI”) scans at 12 months post-injection of OPC1 showed no evidence of adverse changes in any of the 25 patients. | |
| ● | Cell Engraftment. All three patients in Cohort 1 and 21 of the 22 patients in Cohorts 2-5 had MRI scans at 12 months consistent with the formation of a tissue matrix at the injury site, which is encouraging evidence that OPC1 cells had engrafted at the injury site and helped to prevent cavitation. | |
| ● | Improved Motor Function. At 12 months, 21 of the 22 patients who were administered either 10 million or 20 million cells of OPC1 (Cohorts 2-5) recovered at least one motor level on at least one side, and seven of the 22 patients recovered two or more motor levels on at least one side. Motor level recovery was based on the upper extremity motor score (“UEMS”), as measured by the International Standards for Neurological Classification of Spinal Cord Injury (“ISNCSCI”). None of these patients saw decreased motor function following administration of OPC1, and patients consistently retained the motor function recovery seen through six months or saw further motor function recovery from six to 12 months. |
In November 2019, we provided an update on the SCiStar trial that highlighted, among other things:
| ● | Positive Safety Profile. For the 21 SCiStar trial patients who had follow-up visits at 24 months post-injection of OPC1, MRI scans showed no evidence of adverse changes, and none of the patients had a decline in their motor function from their 12-month follow-up visit. There were no unexpected serious adverse events to date in any of these patients. | |
| ● | Improved Motor Function. All 3 Cohort 1 patients continued to be stable 2-4 years out post treatment. At 24 months, five of the six Cohort 2 patients recovered at least two motor levels on at least one side, and one Cohort 2 patient recovered three motor levels, which has been maintained through that patient’s 36-month follow-up visit. Motor level recovery was based on the UEMS as measured by the ISNCSCI. |
In November 2020, the formal Clinical Study Report for the SCiStar study with the above supporting data was submitted to the FDA.
The FDA designated OPC1 as a Regenerative Medicine Advanced Therapy (“RMAT”), for the treatment of acute SCI and granted it Orphan Drug Designation, which includes the ability for increased interfacing with the FDA during clinical development, and a pathway to possible market exclusivity.
| 10 |
In 2019, we transferred all cGMP manufacturing processes, including the establishment of cell banks and the OPC1 process development and manufacturing for clinical studies, to our cell therapy manufacturing facility in Jerusalem, Israel. Improvements to the manufacturing process were completed in 2020 and include enhancements to the production process to ensure robust, controlled reproducible and commercially viable scale, and purity of OPC1. We also developed a thaw and inject formulation of OPC1 to facilitate logistics and handling at the point of care with the elimination of the dose preparation at the clinical site. An information amendment describing the new process, an improved analytical plan, and a proposed comparability plan has been filed with FDA. A meeting with the FDA is planned during the second half of 2021 to discuss our manufacturing improvements and the further development of OPC1 in SCI to best set the program up for success moving forward. Concurrently, we have announced a new partnership for the introduction of a novel delivery device for OPC1. Preliminary assessment of prototypes revealed promising compatibility with OPC1 product while simplifying the surgical procedure by providing surgeons with an instrument that is small, simple to use and would not require stopping the patient’s ventilator to perform the injection, allowing far more flexibility for accurate delivery to the injury site. We intend to complete development activities in the first half of 2021, then discuss with FDA the introduction of the new delivery device in our IND if supported by the collected data. We continue work to expand our partnerships with SCI advocacy and support organizations to support their mission to accelerate stem cell treatments to patients with unmet medical needs and fast-track the development of the most promising stem cell technologies.
VAC2
VAC2 is our lead product candidate for the treatment of cancer. Cancer afflicts millions worldwide and is one of the largest unmet clinical needs with current treatment options providing limited efficacy and a wide range of debilitating side effects. To provide a more effective and targeted treatment, we are developing VAC2 as an allogeneic, or non-patient specific, cancer vaccine candidate designed to stimulate patient immune responses to an antigen hTERT, which is commonly expressed in cancerous cells but not in normal adult cells. VAC2, is produced by our pluripotent cell technology using a directed differentiation method, and is comprised of a population of mature dendritic cells to which the hTERT antigen was introduced. As the most potent type of antigen presenting cell in the body, dendritic cells instruct our body’s immune system to attack and eliminate harmful pathogens and unwanted cells. To target cancerous cells, VAC2 is engineered to express the tumor-selective antigen telomerase, which is found in over 85% of all cancers. The tumor antigen is loaded exogenously into the dendritic cells. The VAC1 autologous program, which preceded VAC2, serves as an effective and encouraging proof of concept behind our approach to dendritic cell vaccines targeting telomerase, which is the backbone of the VAC2 program.
Using pluripotent cells as the starting material for VAC2 production adds several additional advantages to this therapeutic candidate. Compared to technologies that rely on the use of a patient’s own blood, our pluripotent cell technology provides a scalable system for production of a large number of vaccine doses in a single lot, lower manufacturing costs, greater product consistency, and more notably, off-the-shelf availability to provide broader and immediate access to patients. In addition, we believe that as an allogeneic therapy, VAC2 has the potential to stimulate a more robust immune response through an adjuvant effect resulting from the partial immune mismatch between the VAC2 cells and patients receiving the therapy. We believe that VAC2 can be used as a platform technology that can be modified to carry any antigen, including patient-specific tumor neo-antigens.
In September 2014, Asterias initiated clinical development of VAC2 by entering into a Clinical Trial and Option Agreement (the “CRUK Agreement”) with Cancer Research UK (“CRUK”) and Cancer Research Technology Limited (“CRT”), a wholly owned subsidiary of CRUK, under which CRUK agreed to fund Phase 1 clinical development of VAC2 in non-small cell lung cancer. CRUK is responsible, at its own cost, for manufacturing clinical grade VAC2 and for carrying out the Phase 1 clinical trial of VAC2. Patient enrollment began in June 2018 and six patients have now completed dosing in the initial aspect of the trial.
In May 2020, Lineage and its wholly owned subsidiary Asterias entered into a Second Amendment to Clinical Trial and Option Agreement (the “CTOA Amendment”) with CRUK and CRT, which amends the Clinical Trial and Option Agreement entered into between Asterias, CRUK and CRT dated September 8, 2014, as amended September 8, 2014. Pursuant to the CTOA Amendment, Lineage assumed all obligations of Asterias and exercised early its option to acquire data generated in the Phase 1 clinical trial of VAC2 in non-small cell lung cancer being conducted by CRUK. CRUK will continue conducting the VAC2 study.
| 11 |
Lineage and CRT effectuated the option by simultaneously entering into a license agreement (the “License Agreement”) pursuant to which Lineage agreed to pay the previously agreed signature fee of £1,250,000 (approximately $1.6 million). In consideration of Lineage’s agreement to exercise the option prior to completion of the study, the parties agreed to defer the signature fee as follows: £500,000 in September 2020, £500,000 in January 2021 and £250,000 in April 2021. For the primary licensed product for the first indication, the License Agreement provides for milestone fees of up to £8,000,000 based upon initiation of a Phase 3 clinical trial and the filing for regulatory approval and up to £22,500,000 in sales-based milestones payments. Additional milestone fees and sales-based milestone payments would be payable for other products or indications, and mid-single-digit royalty payments are payable on sales of commercial products.
We completed the transfer of all cGMP manufacturing processes, including the establishment of cell banks and the VAC2 process development and manufacturing for clinical studies, to our cell therapy manufacturing facility in Jerusalem, Israel. In 2021, we will focus on updating and optimizing the manufacturing process for VAC to ensure reliable supply for future clinical studies and possible commercial development. An improved VAC manufacturing process will be the subject of a key interaction with FDA in the future to introduce VAC in an IND.
The allogeneic VAC2 program was preceded by the autologous VAC1 program which isolated dendritic cells from a patient’s own blood, modified those cells to stimulate immune responses to telomerase and then administered those cells back to the patient as a therapeutic modality. VAC1 was studied for the treatment of acute myeloid leukemia, the most common form of acute leukemia in adults. A Phase 2 clinical trial of VAC1 demonstrated that it successfully manufactured and released in 24 out of the 33 patients enrolled in the trial. Twenty-one patients received VAC1 in the trial, including 19 in clinical remission and two in early relapse. VAC1 was found to have a favorable safety and tolerability profile. Asterias performed follow-up data collection on the 19 patients treated while in complete remission to determine the long-term effects of the VAC1 administration on remission duration and disease-free survival.
VAC1 utilized an autologous approach where the cellular vaccine needs to be created specifically for each patient. This results in a longer time prior to administration of therapy as compared to the allogeneic approach of the VAC2 program, which is disadvantageous in advanced cancer patients given the rapidity of disease progression. The VAC1 autologous program which preceded VAC2 serves as an effective and encouraging proof of concept behind our approach to dendritic cell vaccines targeting telomerase, which is the backbone of the VAC2 program.
Research Programs
Vision restoration
In 2017, we expanded our ophthalmology portfolio by acquiring exclusive global rights to technology that allows the generation of three-dimensional human retinal tissue derived from human pluripotent cells. This tissue contains all the cell types and layers of the human retina and has shown evidence of functional integration in proof of concept animal models for advanced retinal degeneration. The technology is being developed to potentially treat or prevent a variety of retinal degenerative diseases and injuries. In 2017, the National Institutes of Health (“NIH”) awarded us a grant of up to $1.6 million to further develop this innovative, next generation vision restoration program for retinal diseases and injuries, which severely impact the quality of life for millions of people who have limited treatment options. In 2019, we received an additional grant of $0.7 million to continue work on this program. We completed work under this grant in 2020 and submitted final reports to the NIH.
In 2020, the Israeli Innovation Authority approved a budgeted grant of approximately $0.6 million for us to manufacture novel retinal implants aimed to treat patients with severe retinal impairment such as retinitis pigmentosa. We are eligible for 60% reimbursement of our costs under this grant. This program allows us to combine our knowledge in manufacturing RPE cells and photoreceptors with 3D printing technology.
Demyelination
OPC1 exhibits multiple reparative properties that may have broad applicability to neurological injury and disease, particularly as a treatment for demyelination. Past research efforts investigated the potential development of OPC1 as a candidate treatment for certain forms of ischemic stroke and multiple sclerosis (“MS”), two severely debilitating conditions for which demyelination is a central component to their pathology.
| 12 |
To develop OPC1 as a treatment for MS, initial proof-of-concept efficacy data has been demonstrated in collaboration with Yale University using a non-human primate model of MS. Results of this study showed OPC1 engraftment that was associated with substantial remyelination of the lesioned primate spinal cord up to five months post-treatment. Subsequently, we initiated a collaboration with University of California Irvine to assess OPC1 efficacy in additional mouse models of MS that better recapitulate the autoimmune components of the disease. Preliminary results indicated that in addition to OPC1’s capacity to remyelinate the lesioned spinal cord, the cells may also help stimulate proliferation of a distinct class of immune cells known as regulatory T cells that can help reduce or eliminate autoimmunity.
For ischemic stroke, initial proof-of-concept efficacy data for OPC1 has been demonstrated in a collaborative study with the University of California Los Angeles using a mouse model of white matter ischemic stroke. Results of this study demonstrated that within the stroke injury site, OPC1 cells engrafted, reduced lesion formation and inflammation, and increased myelination, culminating in improved functional recovery. A second preclinical study was completed in collaboration with the University of South Florida to test two different doses of OPC1 in a rat model of ischemic subcortical and white matter stroke. Results from this study demonstrated the ability of OPC1 to impact the restoration of motor function in a rat model of white matter stroke. Further, histological assessments showed a treatment-associated reduction in stroke lesion size, including in the white matter, as well as reduced inflammation and sustained OPC1 engraftment in the injured brain.
While we are not actively pursuing OPC1 for MS and ischemic stroke at this time, we may use the results of these studies to seek additional funding and guide further preclinical development of OPC1 for these or other conditions of demyelination.
Products for Other Indications
We also have rights to intellectual property applicable to other indications such as for producing cardiomyocytes, pancreatic islet cells, hepatocytes, chondrocytes, osteoblasts and other cell types for which development of new therapies represent significant commercial opportunities. We may elect to pursue these or other programs at any time.
Other Products
We also have rights to HyStem, a patented biomaterial that mimics naturally occurring extracellular matrix, the structural network of molecules surrounding cells in organs and tissues essential to cellular function and tissue structure. HyStem may be useful as a scaffold for cell replacement and retention. We sold HyStem-related assets and licensed the applicable technology in late 2019, but retained the rights for other uses, including for Renevia, our facial aesthetics product, which received a Conformité Européenne (CE) Mark in September 2019.
Investments and subsidiaries:
The following tables show the companies in which we have a direct or indirect ownership, their respective principal fields of business, our percentage ownership as of March 5, 2021, and the country where their principal business is located:
Investments:
| Company | Field of Business | Lineage Ownership | Country | |||||
| OncoCyte Corporation(1) | Cancer diagnostics | ~1 | % | USA | ||||
| Hadasit Bio-Holdings Ltd.(1) | Owns a portfolio of R&D based companies | <2 | % | Israel |
| 13 |
Significant subsidiaries:
| Company | Field of Business | Lineage Ownership | Country | |||||
| Cell Cure Neurosciences Ltd. | Development and manufacturing of Lineage’s cell replacement platform technology | 99 | %(2) | Israel | ||||
| Asterias Biotherapeutics, Inc.(3) | Cell based therapeutics to treat neurological conditions | 100 | % | USA | ||||
| ES Cell International Pte. Ltd(4) | Research and clinical grade cell lines | 100 | % | Singapore | ||||
| OrthoCyte Corporation(4)(5) | Research in orthopedic diseases and injuries | 99.8 | % | USA | ||||
| (1) | These are publicly traded companies. See Notes to Consolidated Financial Statements: Note 4. Equity Method of Accounting for Common Stock of OncoCyte, at Fair Value. |
| (2) | Includes shares owned by Lineage and ES Cell International Pte. Ltd. (“ESI”). |
| (3) | Asterias was acquired by Lineage in March 2019. See Notes to Consolidated Financial Statements: Note 3. Asterias Merger. |
| (4) | The operating activities and fields of business listed under these subsidiaries are conducted primarily by Lineage as the parent company. |
| (5) | OrthoCyte Corporation (“OrthoCyte”) adopted a stock option plan under which it may issue up to 4,000,000 shares of its common stock to officers, directors, employees, and consultants of OrthoCyte and Lineage employees, including officers. As of December 31, 2020, no options to purchase OrthoCyte common stock were outstanding. |
Patents and Trade Secrets
We seek to protect and rely on our proprietary cell-based therapy platform and associated development and manufacturing capabilities and derived product candidates through a variety of methods, including seeking and maintaining patents intended to cover our products and compositions, their methods of use and processes for their manufacture, our platform technologies and any other inventions that are commercially important to the development of our business. We also rely on contractual obligations with employees and third parties to protect our proprietary rights. For example, in addition to protecting our proprietary rights with patents, we rely on unpatented trade secrets, improvements, know-how and innovation, and we take steps necessary to protect these rights, including through confidentiality agreements with our corporate partners, employees, consultants and vendors. We have sought, and intend to continue to seek, appropriate patent protection for important and strategic components of our proprietary technologies by filing patent applications in the U.S. and internationally. We may also file additional patent applications, when appropriate, to cover improvements on our clinical products, clinical product candidates, and related technologies. There are no assurances that any of our intellectual property rights will guarantee complete or adequate protection or market exclusivity for our products and product candidates. We also enter into collaborative and other similar arrangements with third parties, such as license agreements, to in-license and/or out-license intellectual property rights. Our financial success will be dependent, in part, on our ability to obtain rights to commercially valuable patents, to protect and enforce our intellectual property rights and to operate without infringing any intellectual property rights of others. From time to time, we assess our patents and pending applications covering our products and product candidates. If we determine that any patents or patent applications no longer provide adequate or necessary protection, we may transfer or abandon such patents and patent applications to avoid incurring unnecessary costs.
We own or license, directly or through our subsidiaries, several patent families that include hundreds of U.S. and international patents and patent applications. We cannot be certain that issued patents will be enforceable or provide adequate protection or that pending applications will result in issued patents.
OpRegen
We and our subsidiary, Cell Cure, have rights to issued U.S. and international patents and pending patent applications covering OpRegen. The issued patents have expiration dates ranging from 2028 to 2036. The pending applications if issued, will have estimated expiration dates ranging from 2028 to 2041. These U.S. and international issued patents and pending applications also include those in-licensed from Hadasit Medical Research Services and Development Ltd. (“Hadasit”), the commercial arm and a wholly owned subsidiary of Hadassah Medical Organization. We also solely own pending U.S. and Patent Cooperation Treaty (“PCT”) patent applications relating to cryopreserving the cell population and then shipping it to the clinical trial site so the cells can be immediately thawed and delivered to the patient without further processing. The U.S. patent applications, and any filed international patent applications based on the PCT applications, if issued, will have estimated expiration dates in 2038.
| 14 |
Cell Cure was a party to two pending opposition proceedings in the European Patent Office (“EPO”) involving EP Patent Numbers 2147094 (issued 08-Oct-2014) and 2554661 (issued 19-Nov-2014), both entitled, “Stem Cell-Derived Retinal Pigment Epithelial Cells”. The oral proceedings took place on March 16, 2017 and March 17, 2017, respectively. Both patents were upheld by the EPO and the patents issued as amended during the opposition proceedings. Both patents cover OpRegen until 2028.
OPC1
We have numerous U.S. and international issued patents and pending patent applications that are relevant to neural cells, such as oligodendrocyte progenitor cells, including patent families acquired from Geron Corporation (“Geron”) that are directed to the differentiation of pluripotent stem cells, including human embryonic stem (“hES”) cells, into various neural cell types, as well as various culture and purification methods. These U.S. and international issued patents and pending patent applications also include those in-licensed from the Regents of the University of California. Additionally, there are four patent families with pending patent applications owned by us directed to improved methods of producing oligodendrocyte progenitor cells, oligodendrocyte progenitor cell compositions and methods of treatment of spinal cord injury using oligodendrocyte progenitor cells. There is also a patent family directed to improved methods of producing oligodendrocyte progenitor cells, oligodendrocyte progenitor cell compositions and methods for the treatment of stroke using oligodendrocyte progenitor cells which is jointly owned with the Regents of the University of California. The expiration dates of the patents and pending patent applications acquired from Geron and in-licensed from the Regents of the University of California range from 2023 to 2036. The estimated expiration dates of the four patent families with pending applications owned by us range from 2036 to 2040. The commercial success of OPC1 depends, in part, upon our ability to exclude competition for this product with the existing patent portfolio, regulatory exclusivity, undisclosed know-how and/or trade secrets, or a combination of these barriers to entry.
VAC1 and VAC2
We have numerous U.S. and international issued patents and pending patent applications that are relevant to dendritic cells, including patent families acquired from Geron or in-licensed from third parties that are directed to the differentiation of pluripotent stem cells, including hES cells, into hematopoietic progenitor cells and immature and mature dendritic cells. In addition, these patent rights include a patent family with claims directed to immunogenic compositions comprising antigen-presenting dendritic cells and methods of eliciting an anti-telomerase immune response in a subject by administering to the subject such compositions. The expiration dates of the patents, and the estimated expiration dates of the pending applications, acquired from Geron or in-licensed to us range from 2022 to 2041. The commercial success of VAC1 and VAC2 products depends, in part, upon our ability to exclude competition in these products with this patent portfolio, regulatory exclusivity, undisclosed know-how and/or trade secrets, or a combination of these barriers to entry.
Other Patents and Patent Applications
We have U.S. and international issued patents and pending patent applications related to producing cardiomyocytes, pancreatic islet cells, hepatocytes, chondrocytes and osteoblasts. The expiration dates of these patents and pending patent applications range from 2020 to 2032. In addition, we have U.S. and international issued patents and pending patent applications related to suspension cultures and feeder-free cultures for culturing and proliferating pluripotent stem cells. The expiration dates for these patents and pending patent applications range from 2021 to 2026.
We also have U.S. and international issued patents and pending applications covering Renevia, include those in-licensed from the University of Utah Research Foundation (“UURF”) having expiration dates ranging from 2023 to 2027, and a pending patent application in Europe having an estimated expiration date of 2024. We also solely own pending U.S. and European patent applications filed in 2018 that, if issued, will have estimated expiration dates in 2038.
| 15 |
General Risks Related to Obtaining and Enforcing Patent Protection
Because patent applications are confidential until a patent is issued, we may not know if our competitors have filed patent applications for technology covered by our pending applications or if we were the first to invent or first to file an application directed toward the technology that is the subject of our patent applications. Competitors may have filed patent applications or received patents and may obtain additional patents and proprietary rights that block or compete with our products. In addition, if competitors file patent applications covering our technology, we may have to participate in interference/derivation proceedings or litigation to determine the right to a patent. Litigation and interference/derivation proceedings are unpredictable and expensive, such that, even if we are ultimately successful, our results of operations may be adversely affected by such events. Accordingly, there is a risk that any patent applications that we file and any patents that we hold or later obtain could be challenged by third parties and be declared invalid in view of third party patent applications and/or patents. Litigation, interferences, oppositions, inter partes reviews or other proceedings are, have been and may in the future be necessary in some instances to determine the validity and scope of certain of our proprietary rights, and in other instances to determine the validity, scope or non-infringement of certain patent rights claimed by third parties to be pertinent to the manufacture, use or sale of our products. We may also face challenges to our patent and regulatory protections covering our products by third parties, including manufacturers of generics and biosimilars that may choose to launch or attempt to launch their products before the expiration of our patent or regulatory exclusivity. Litigation, interference, oppositions, inter partes reviews, administrative challenges or other similar types of proceedings are unpredictable and may be protracted, expensive and distracting to management. The outcome of such proceedings could adversely affect the validity and scope of our patent or other proprietary rights, hinder our ability to manufacture and market our products, require us to seek a license for the infringed product or technology or result in the assessment of significant monetary damages against us that may exceed any amounts that we may accrue on our financial statements as a reserve for contingent liabilities. An adverse determination in a judicial or administrative proceeding or a failure to obtain necessary licenses could prevent us from manufacturing or selling our products. Furthermore, payments under any licenses that we are able to obtain would reduce our profits derived from the covered products and services.
The enforcement of patent rights often requires litigation against third-party infringers, and such litigation can be costly to pursue. Even if we succeed in having new patents issued or in defending any challenge to issued patents, there is no assurance that our patents will be comprehensive enough to provide us with meaningful patent protection against our competitors.
Employees
As of December 31, 2020, we had 55 employees, of which 20 were Lineage employees and 35 were employees of Cell Cure in Israel and of which 49 were employed on a full-time basis and six were employed on a part-time basis. Ten employees hold Ph.D. degrees in one or more fields of science. None of our employees are covered by a collective bargaining agreement.
Manufacturing
We maintain an innovative cell therapy manufacturing facility in the Bio Park on the campus of the Hadassah University Hospital in Jerusalem, Israel. The facility includes process development laboratories and a state-of-the-art, cGMP manufacturing facility. It is designed and equipped to enable simultaneous cGMP processes and to produce a range of cell therapy products for human use in clinical trials as well as at a scale suitable for commercial launch. All cGMP manufacturing processes, including cell banks and product manufacturing for our cell therapy product candidates are conducted in this facility.
We obtain key components required for the manufacture of our cell therapy product candidates from third-party manufacturers and suppliers, which include, in some instances, sole source manufacturers and suppliers. We do not currently have long-term commitments or supply agreements in place to obtain certain key components used in the manufacture of our cell therapy product candidates.
Licensed Technology and Product Development Agreements
Lineage has obtained the right to use technology that we believe has great potential in our product development efforts, and that may be useful to other companies that are engaged in the research and development of products for human therapeutic and diagnostic use.
| 16 |
Second Amendment to Clinical Trial and Option Agreement and License Agreement with Cancer Research UK
On May 6, 2020, Lineage and its wholly owned subsidiary Asterias entered into a Second Amendment to Clinical Trial and Option Agreement (the “CTOA Amendment”) with Cancer Research UK (“CRUK”) and Cancer Research Technology Limited (“CRT”), which amends the Clinical Trial and Option Agreement entered into between Asterias, CRUK and CRT dated September 8, 2014, as amended September 8, 2014. Pursuant to the CTOA Amendment, Lineage assumed all obligations of Asterias and exercised early its option to acquire data generated in the Phase 1 clinical trial of VAC2 in non-small cell lung cancer being conducted by CRUK. CRUK is continuing to conduct the VAC2 study.
Lineage and CRT effectuated the option by simultaneously entering into a license agreement (the “CRT License Agreement”) pursuant to which Lineage agreed to pay the previously agreed signature fee of £1,250,000 (approximately $1.6 million). In consideration of Lineage’s agreement to exercise the option prior to completion of the study, the parties agreed to defer the signature fee as follows: £500,000 in September 2020, £500,000 in January 2021 and £250,000 in April 2021. For the primary licensed product for the first indication, the CRT License Agreement provides for milestone fees of up to £8,000,000 based upon initiation of a Phase 3 clinical trial and the filing for regulatory approval and up to £22,500,000 in sales-based milestones payments. Additional milestone fees and sales-based milestone payments would be payable for other products or indications, and mid-single-digit royalty payments are payable on sales of commercial products.
Either party may terminate the CRT License Agreement for the uncured material breach of the other party. CRT may terminate the CRT License Agreement in the case of Lineage’s insolvency or if Lineage ceases all development and commercialization of all products under the CRT License Agreement.
Hadasit Research and License Agreement
In June 2017, Cell Cure entered into a Second Amended and Restated License Agreement with Hadasit (the “Hadasit License Agreement”). Pursuant to the Hadasit License Agreement, Hadasit granted Cell Cure an exclusive, worldwide, royalty-bearing license (with the right to grant sublicenses) in its intellectual property portfolio of U.S. and international issued patents and pending patent applications relevant to materials and technology related to human stem cell derived photoreceptor cells and RPE cells, to use, commercialize and exploit any part thereof, in any manner whatsoever in the fields of the development and exploitation of: (i) human stem cell derived photoreceptor cells, solely for use in cell therapy for the diagnosis, amelioration, prevention and treatment of eye disorders; and (ii) human stem cell derived RPE cells, solely for use in cell therapy for the diagnosis, amelioration, prevention and treatment of eye disorders. This intellectual property licensed includes patents and pending applications having expiration dates, and estimated expiration dates, respectively, ranging from 2025 to 2028. Cell Cure and Hadasit also jointly own U.S. and international issued patents and patent applications directed to methods of selecting RPE cells, which patents and patent applications will expire in 2033.
Pursuant to the Hadasit License Agreement, Cell Cure paid a small one-time lump sum payment for reimbursement of intellectual property related expenses and will pay a royalty in the mid-single digits of net sales from sales of licensed intellectual property by any invoicing entity and a royalty of 21.5% on sublicensing receipts. In addition, Cell Cure will pay Hadasit an annual minimal non-refundable royalty, which will become due and payable the first January 1 following the completion of services to Cell Cure by a research laboratory.
Cell Cure agreed to pay Hadasit non-refundable milestone payments upon the recruitment of the first patient for the first Phase 2b clinical trial, upon the enrollment of the first patient in the first Phase 3 clinical trials, upon delivery of the report for the first Phase 3 clinical trials, upon the receipt of an NDA or marketing approval in the EU, whichever is the first to occur, and upon the first commercial sale in the United States or EU, whichever is the first to occur. Such milestones, in the aggregate, may be up to $3.5 million. As of December 31, 2020, Cell Cure had not accrued any of these milestone payments.
The Hadasit License Agreement terminates upon the expiration of Cell Cure’s obligation to pay royalties for all licensed products, unless earlier terminated. In addition, the Hadasit License Agreement may be terminated by (i) Hadasit if, among other reasons, Cell Cure fails to continue the clinical development of the licensed intellectual property or fails to take actions to commercialize or sell the licensed intellectual property over any consecutive 12-month period, and (ii) by either party for: (a) a material breach which remains uncured following a cure period; or (b) the granting of a winding-up order in respect of the other party, or upon an order being granted against the other party for the appointment of a receiver or a liquidator in respect of a substantial portion of such other party’s assets. The Hadasit License Agreement also contains customary indemnification obligations of Cell Cure.
| 17 |
License Agreement with University of California
We are party to an exclusive license agreement with The Regents of the University of California dated February 20, 2003 (the “UC License Agreement”) for U.S. and international issued patents and pending patent applications covering a method for directing the differentiation of pluripotent cells to glial-restricted progenitor cells that generate pure populations of oligodendrocytes for remyelination and treatment of spinal cord injury. Under the UC License Agreement, we have an exclusive worldwide license under such patents, including the right to grant sublicenses, to create products for biological research, drug screening, and human therapy using the licensed patents. These issued patents and pending applications have expiration dates ranging from 2023 to 2024.
Under the UC License Agreement, we will pay the university a royalty of 1% from sales of products that are covered by the licensed patent rights, and a minimum annual royalty of $5,000 starting in the year in which the first sale of a product covered by any licensed patent rights occurs and continuing for the life of the applicable patent right under the agreement. Under certain conditions, we will pay the university 7.5% of any proceeds, excluding debt financing and equity investments, and certain reimbursements, that we receive from sublicensees.
The UC License Agreement terminates on the expiration of the last-to-expire of the university’s issued licensed patents. If no further patents covered by the UC License Agreement are issued, it will terminate in 2024. The university may terminate the UC License Agreement if we breach it, and we can terminate with 60 days’ notice.
WARF Agreements
We have rights to certain U.S and international issued patents, pending patent applications and stem cell lines with the Wisconsin Alumni Research Foundation (“WARF”) under a Commercial License and Option Agreement entered into between Lineage and WARF in January 2008 and a Non-Exclusive License Agreement entered into between Asterias and WARF in October 2013 (collectively, the “WARF Agreements”).
Under the WARF Agreements, we have a worldwide non-exclusive license under certain WARF patents and WARF-owned primate (including human) stem cell lines covered by such patents for use in internal research, and to make, use and sell products that are used as research tools and products that are discovered or developed through our internal research using such patents and stem cells. We paid upfront license fees and have agreed to additional payments upon the attainment of specified clinical development milestones, royalties on sales of commercialized products, and, subject to certain exclusions, a percentage of any payments that we may receive from any sublicenses that we may grant to use the licensed patents or stem cell lines.
The WARF Agreements will terminate with respect to licensed patents upon the expiration of the last licensed patent to expire and with respect to licensed cell lines until terminated by a party. We may terminate the WARF Agreements at any time with prior written notice, and WARF may terminate the WARF Agreements upon a breach. We have agreed to indemnify WARF and certain other designated affiliated entities from liability arising out of or relating to the death or injury of any person or damage to property due to the sale, marketing, use or manufacture of products that are covered by the licensed patents, licensed stem cell lines or inventions or materials developed or derived from the licensed patents or stem cell lines.
Royalty Agreement with Geron
In connection with Asterias’s acquisition of Geron’s stem cell assets, in October 2013, we entered into a royalty agreement with Geron (the “Royalty Agreement”) pursuant to which we agreed to pay Geron a 4% royalty on net sales (as defined in the Royalty Agreement) by us or any of our affiliates or sales agents of any products that we develop and commercialize that are covered by the patents Geron contributed to us. In the case of sales of such products by a person other than us or one of our affiliates or sales agents, we will be required to pay Geron 50% of all royalties and cash payments received by us or by our affiliate in respect of a product sale. Royalty payments will be subject to proration in the event that a product covered by a patent acquired from Geron is sold in combination with another product that is not covered by a patent acquired from Geron. The Royalty Agreement will terminate at the expiration or termination date of the last issued patent contributed by Geron under the Royalty Agreement. We estimate that the latest patent expiration date will be in 2032.
| 18 |
Government Regulation
Government authorities at the federal, state and local level, and in other countries, extensively regulate among other things, the development, testing, manufacture, quality, approval, safety, efficacy, distribution, labeling, packaging, storage, record keeping, marketing, import/export and promotion of drugs, biologics, and medical devices. Authorities also heavily regulate many of these activities for human cells, tissues, and cellular and tissue-based products (“HCT/Ps”).
FDA and Foreign Regulation of Therapeutic Products
The FDA and foreign regulatory authorities will regulate our proposed products as drugs, biologics or medical devices, depending upon such factors as the use to which the product will be put, the chemical composition, and the interaction of the product with the human body. In the United States, the FDA regulates drugs and biologics under the Federal Food, Drug and Cosmetic Act (“FDCA”), the Public Health Service Act (“PHSA”), and implementing regulations. In addition, establishments that manufacture human cells, tissues, and HCT/Ps are subject to additional registration and listing requirements, including current good tissue practice regulations. Certain cell therapy proposed products will be reviewed by the FDA staff in its Center for Biologics Evaluation and Research Office of Cellular, Tissue and Gene Therapies.
Our domestic human drug and biologic products will be subject to rigorous FDA review and approval procedures. After testing in animals to evaluate the potential efficacy and safety of the product candidate, an investigational new drug (“IND”) submission must be made to the FDA to obtain authorization for human testing. Extensive clinical testing, which is generally done in three phases, must then be undertaken to demonstrate optimal use, safety, and efficacy of each product in humans. Each clinical trial is conducted under the auspices of an independent Institutional Review Board (“IRB”). The IRB will consider, among other things, ethical factors, the safety of human subjects, and the possible liability of the institution.
Phase 1 clinical trials are conducted in a small number of healthy volunteers or volunteers with the target disease or condition to assess safety. Phase 2 clinical trials are conducted with groups of patients afflicted with the target disease or condition in order to determine preliminary efficacy, optimal dosages and expanded evidence of safety. In some cases, an initial trial is conducted in diseased patients to assess both preliminary efficacy and preliminary safety, in which case it is referred to as a Phase 1/2 clinical trial. Phase 3 clinical trials are large-scale, multi-center, comparative trials and are conducted with patients afflicted with the target disease or condition in order to provide enough data to demonstrate the efficacy and safety required by the FDA. The FDA closely monitors the progress of each of the three phases of clinical testing and may, at its discretion, re-evaluate, alter, suspend or terminate the clinical trial based upon the data which have been accumulated to that point and its assessment of the risk/benefit ratio to the intended patient population. All adverse events must be reported to the FDA. Monitoring of all aspects of the trial to minimize risks is a continuing process.
No action can be taken to market any therapeutic product in the U.S. until an appropriate New Drug Application (“NDA”) or Biologics License Application (“BLA”) has been approved by the FDA. Submission of the application is not a guarantee that the FDA will find it complete and accept it for filing. If an application is accepted for filing, following the FDA’s review, the FDA may grant marketing approval, request additional information or deny the application by way of a Complete Response Letter if it determines that the application does not provide an adequate basis for approval. FDA regulations also restrict the export of therapeutic products for clinical use prior to FDA approval. Before approving a BLA, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications including gene therapy products (“GTPs”) to the extent applicable. These are FDA regulations and guidance documents that govern the methods used in, and the facilities and controls used for, the manufacture of HCT/Ps. The primary intent of the GTP requirements is to ensure that cell and tissue-based products are manufactured in a manner designed to prevent the introduction, transmission and spread of communicable disease. FDA regulations also require HCT/P establishments to register and list their HCT/Ps with the FDA and, when applicable, to evaluate donors through screening and testing. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure that the clinical trials were conducted in compliance with IND trial requirements and GCP requirements. To maintain compliance with CGMPs, GTPs, and GCPs, an applicant must incur significant expenditure of time, money and effort in the areas of training, record keeping, production, and quality control.
| 19 |
To date, the FDA has not granted marketing approval to any pluripotent stem-based therapeutic products and it is possible that the FDA or foreign regulatory agencies may subject our product candidates to additional or more stringent review than drugs or biologics derived from other technologies.
The FDA offers several programs to expedite development of products that treat serious or life-threatening illnesses and that provide meaningful therapeutic benefits to patients over existing treatments. A drug is eligible for designation as an RMAT if: the drug is a regenerative medicine therapy, which is defined as a cell therapy, therapeutic tissue engineering product, human cell and tissue product or any combination product using such therapies or products, except for those regulated solely under certain other sections; the drug is intended to treat, modify, reverse or cure a serious or life-threatening disease or condition; and preliminary clinical evidence indicates that the drug has the potential to address unmet medical needs for such disease or condition. Some of our current and future products may be eligible for RMAT designation.
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biologic intended to treat a rare disease or condition, which is a disease or condition that affects fewer than 200,000 individuals in the United States, or if it affects more than 200,000 individuals in the United States, there is no reasonable expectation that the cost of developing and making available a drug or biologic for this type of disease or condition will be recovered from sales in the United States for that drug or biologic. Orphan drug designation must be requested before submitting a BLA. After the FDA grants orphan drug designation, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. The orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review or approval process.
If a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications, including a full BLA, to market the same biologic for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. Orphan drug exclusivity does not prevent FDA from approving a different drug or biologic for the same disease or condition, or the same drug or biologic for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the BLA application fee. A designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. In addition, exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
Combination Products
If we develop any products that are used with medical devices, they may be considered combination products, which are defined by the FDA to include products comprised of two or more regulated components or parts such as a biologic and a device. For example, our HyStem hydrogel products may be used to administer one or more pluripotent stem cell-based therapy products. When regulated independently, biologics and devices each have their own regulatory requirements. However, the regulatory requirements for a combination product comprised of a biologic administered with a delivery device can be more complex, because in addition to the individual regulatory requirements for each component, additional combination product regulatory requirements may apply. The Office of Combination Products at the FDA coordinates the review of such products and determines the primary mode of action of a combination product. The definition and regulatory requirements for combination products may differ significantly among countries in which we may seek approval of our product candidates.
FDA Regulation of Manufacturing
The FDA regulates the manufacturing process of pharmaceutical products, human tissue and cell products, and medical devices, requiring that they be produced in compliance with cGMP. See “Manufacturing.” The FDA regulates and inspects equipment, facilities, laboratories and processes used in the manufacturing and testing of products prior to providing approval to market products. If after receiving approval from the FDA, a material change is made to manufacturing equipment or to the location or manufacturing process, additional regulatory review may be required. The FDA also conducts regular, periodic visits to re-inspect the equipment, facilities, laboratories and processes of manufacturers following an initial approval. If, as a result of those inspections, the FDA determines that equipment, facilities, laboratories or processes do not comply with applicable FDA regulations and conditions of product approval, the FDA may seek civil, criminal or administrative sanctions and/or remedies against the manufacturer, including suspension of manufacturing operations. Issues pertaining to manufacturing equipment, facilities or processes may also delay the approval of new products undergoing FDA review.
| 20 |
FDA Regulation of Advertising and Product Promotion
The FDA also regulates the content of advertisements used to market pharmaceutical and biologic products. Claims made in advertisements concerning the safety and efficacy of a product, or any advantages of a product over another product, must be supported by clinical data filed as part of an NDA, a BLA, or an amendment to an NDA or a BLA, and must be consistent with the FDA-approved labeling and dosage information for that product.
Pharmaceutical and biologic products may be promoted only for the approved indications in accordance with the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label may be subject to significant liability. However, physicians may, in their independent medical judgment, prescribe legally available products for off-label uses. The FDA does not regulate the behavior of physicians in their choice of treatments but the FDA does restrict manufacturer’s communications on the subject of off-label use of their products.
Foreign Regulation
Sales of pharmaceutical products outside the U.S. are subject to foreign regulatory requirements that vary widely from country to country. Even if FDA approval has been obtained, approval of a product by comparable regulatory authorities of foreign countries must be obtained prior to the commencement of marketing the product in those countries. The time required to obtain such approval may be longer or shorter than that required for FDA approval.
Federal Funding and State Regulations
Effective July 7, 2009, the National Institutes of Health (“NIH”) adopted guidelines on the use of hES cells in federally funded research. The central focus of the guidelines is to assure that hES cells used in federally funded research are derived from human embryos that were created for reproductive purposes, are no longer needed for this purpose, and are voluntarily donated for research purposes with the informed written consent of the donors. hES cells that were not derived in compliance with the guidelines, are not eligible for use in federally funded research.
The state of California has adopted legislation and regulations that require institutions that conduct stem cell research to notify, and in certain cases obtain approval from, a Stem Cell Research Oversight Committee (“SCRO Committee”) before conducting the research. Under certain California regulations, all hES cell lines used in our research must be acceptably derived. California regulations further require certain records to be maintained with respect to stem cell research and the materials used. Lineage programs that involve the use of stem cells have been reviewed by a SCRO Committee to confirm compliance with federal and state guidelines.
The hES cell lines that we use are all on the NIH registry of lines that have been reviewed and meet standards for federal funding grants. All of our research programs utilize stem cells from established and well-characterized cell lines and which are capable of self-renewal and expansion through normal cellular division (mitosis). Our research programs do not require new tissue or cells from donors of any kind.
Health Insurance Portability and Accountability Act and Other Health Information Privacy and Security Laws
The Health Insurance Portability and Accountability Act (“HIPAA”), as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH”), and their respective implementing regulations impose obligations on “covered entities,” including certain healthcare providers, health plans, and healthcare clearinghouses, as well as their respective “business associates” that create, receive, maintain or transmit individually identifiable health information for or on behalf of a covered entity, and their subcontractors that use, disclose, access, or otherwise process individually identifiable protected health information, with respect to protecting the privacy, security, and transmission of protected health information. HIPAA also regulates standardization of data content, codes and formats used in health care transactions and standardization of identifiers for covered health plans and providers. Penalties for violations of HIPAA regulations include civil and criminal penalties. Additionally, HITECH created four new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in U.S. federal courts to enforce HIPAA and seek attorneys’ fees and costs associated with pursuing federal civil actions. In addition, certain state and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
| 21 |
Federal and State Fraud and Abuse Laws
A variety of federal and state laws prohibit fraud and abuse. These laws are interpreted broadly and enforced aggressively by various state and federal agencies, including the Centers for Medicare & Medicaid Services (“CMS”), the Department of Justice, the Office of Inspector General for HHS, and various state agencies. In addition, the Medicare and Medicaid programs increasingly use a variety of contractors to review claims data and to identify improper payments as well as fraud and abuse. These contractors include Recovery Audit Contractors, Medicaid Integrity Contractors and Zone Program Integrity Contractors. In addition, CMS conducts Comprehensive Error Rate Testing audits, the purpose of which is to detect improper Medicare payments. Any overpayments identified must be repaid unless a favorable decision is obtained on appeal. In some cases, these overpayments can be used as the basis for an extrapolation, by which the error rate is applied to a larger universe of claims, and which can result in even higher repayments.
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting, receiving, or providing remuneration, directly or indirectly, to induce or in return for either the referral of an individual, or the furnishing, recommending, or arranging for the purchase, lease or order of any health care item or service reimbursable, in whole or in part, under a federal health care program. The definition of “remuneration” has been broadly interpreted to include anything of value, including gifts, discounts, credit arrangements, payments of cash, ownership interests and providing anything at less than its fair market value. Recognizing that the federal Anti- Kickback Statute is broad and may prohibit certain common activities within the health care industry, the Office of Inspector General for HHS has issued a series of statutory exceptions and regulatory “safe harbors.” However, these exceptions and safe harbors are drawn narrowly and require strict compliance in order to offer protection from prosecution under the federal Anti-Kickback Statute. Although full compliance with these provisions ensures against prosecution under the federal Anti-Kickback Statute, the failure of a transaction or arrangement to fit within a specific safe harbor does not necessarily mean that the transaction or arrangement is illegal or that prosecution under the federal Anti-Kickback Statute will be pursued. However, conduct and business arrangements that do not fully satisfy all requirements of an applicable safe harbor may result in increased scrutiny by government enforcement authorities and would be evaluated on a case-by-case basis based on a cumulative review of their facts and circumstances. Additionally, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, the “ACA”) codified case law that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act.
The federal civil and criminal false claims laws, including the federal False Claims Act, which can be enforced by private citizens on behalf of the government, through civil whistleblower or qui tam actions, and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent. Pharmaceutical and other health care companies have been prosecuted under these laws for alleged off-label promotion of drugs, purportedly concealing price concessions in the pricing information submitted to the government for government price reporting purposes, and allegedly providing free product to customers with the expectation that the customers would bill federal healthcare programs for the product. As a result of a modification made by the Fraud Enforcement and Recovery Act of 2009, a claim includes “any request or demand” for money or property presented to the U.S. government. In addition, manufacturers can be held liable under the federal False Claims Act even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims.
HIPAA also created new federal crimes, including health care fraud and false statements relating to health care matters. The health care fraud statute prohibits knowingly and willfully executing a scheme to defraud any health care benefit program, including private third-party payers. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for health care benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
| 22 |
The federal Physician Payments Sunshine Act which require certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to CMS information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists, and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Beginning in 2022, applicable manufacturers will also be required to report information regarding payments and other transfers of value provided during the previous year to physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists and anesthesiologist assistants, and certified nurse-midwives.
Many states have laws similar to the federal laws described above and the state laws may be broader in scope and may apply regardless of payor, such as state anti-kickback and false claims laws that may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third party payors, including private insurers, or that apply regardless of payor, state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, state and local laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures, state laws that require the reporting of information related to drug pricing, and state and local laws requiring the registration of pharmaceutical sales representatives.
Additionally, the U.S. Foreign Corrupt Practices Act (“FCPA”) prohibits U.S. corporations and their representatives from offering, promising, authorizing or making payments to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business abroad. The scope of the FCPA includes interactions with certain healthcare professionals in many countries. Other countries have enacted similar anti-corruption laws and/or regulations.
If our operations are found to be in violation of any of the laws described above, or any other governmental regulations that apply to us, we may be subject significant civil, criminal and administrative penalties, including sanctions, damages, disgorgement, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, imprisonment, integrity oversight and reporting obligations, contractual damages, reputational harm, diminished profits and future earnings, and curtailment or restructuring of our operations.
Coverage and Reimbursement
Patients generally rely on third-party payors to reimburse part or all of the costs associated with medical products. Accordingly, market acceptance of medical products can depend on the extent to which third-party coverage and reimbursement is available from government health administration authorities, private healthcare insurers and other healthcare funding organizations. No uniform policy for coverage and reimbursement exists in the United States, and coverage and reimbursement can differ significantly from payor to payor. Decisions regarding whether to cover any of our product candidates, if approved, the extent of coverage and amount of reimbursement to be provided are made on a plan-by-plan basis. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement rates, but also have their own methods and approval process apart from Medicare determinations. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our product candidates to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. Pharmaceutical companies may be required to provide specified rebates or discounts on the products it sells to certain government funded programs, including Medicare and Medicaid, and those rebates or discounts have increased over time. The ACA increased many of these mandatory discounts and rebates required and imposed a new branded prescription pharmaceutical manufacturers and importers fee payable each year by certain pharmaceutical companies and manufacturers.
Outside of the United States, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the EU provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product, or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. Historically, products launched in the EU do not follow price structures of the United States and generally tend to be significantly lower.
| 23 |
Healthcare Reform
The United States and some foreign jurisdictions are considering or have enacted a number of reform proposals to change the healthcare system. There is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by federal and state legislative initiatives, including those designed to limit the pricing, coverage, and reimbursement of pharmaceutical and biopharmaceutical products, especially under government-funded health care programs, and increased governmental control of drug pricing.
In March 2010, the ACA was signed into law, which substantially changed the way healthcare is financed by both governmental and private insurers in the United States, and significantly affected the pharmaceutical industry. The ACA contains a number of provisions of particular import to the pharmaceutical and biotechnology industries, including, but not limited to, those governing enrollment in federal healthcare programs, a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. Since its enactment, there have been judicial, Congressional, and executive branch challenges to certain aspects of the ACA, and we expect there will be additional challenges and amendments to the ACA in the future. For example, the 2020 federal spending package permanently eliminated, effective January 1, 2020, the ACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, also eliminated the health insurer tax. In addition, legislation enacted in 2017, informally known as the Tax Cuts and Jobs Act (the “2017 Tax Act”), among other things, removes penalties for not complying with ACA’s individual mandate to carry health insurance. Since the enactment of the 2017 Tax Act, there have been additional amendments to certain provisions of the ACA. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas, ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the 2017 Tax Act, the remaining provisions of the ACA are invalid as well. Additionally, on December 18, 2019, the U.S. Court of Appeals for the 5th Circuit upheld the District Court ruling that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. The U.S. Supreme Court is currently reviewing the case, although it is unknown when or how the Supreme Court will rule. Accordingly, it is unclear how this decision, future decisions, subsequent appeals, if any, and other efforts to repeal and replace the ACA will impact the ACA.
Moreover, there has recently been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. At the federal level, the Trump administration’s budget proposal for fiscal year 2021 includes a $135 billion allowance to support legislative proposals seeking to reduce drug prices, increase competition, lower out-of-pocket drug costs for patients, and increase patient access to lower-cost generic and biosimilar drugs. Further, the Trump administration released a “Blueprint”, or plan, to lower drug prices and reduce out of pocket costs of drugs that contains additional proposals to increase drug manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products, and reduce the out of pocket costs of drug products paid by consumers. The likelihood of implementation of any of these, or the other Trump administration reform initiatives is uncertain, particularly in light of the new presidential administration. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Further, it is possible that additional governmental action is taken in response to the COVID-19 pandemic.
| 24 |
Major Customers and Sources of Revenues
Major Sources of Revenues
The following table shows our major sources of revenues, as a percentage of total revenues, that were recognized during the years ended December 31, 2020 and 2019:
| Year Ended December 31, | ||||||||
| Sources of Revenues | 2020 | 2019 | ||||||
| NIH grant income | 21.2 | % | 17.5 | % | ||||
| IIA grant income (Cell Cure Neurosciences Ltd, Israel) | 36.5 | % | 40.5 | % | ||||
| Royalties from product sales and licenses fees | 42.3 | % | 34.7 | % | ||||
| Sale of research products | - | % | 7.3 | % | ||||
Geographic Area
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| United States | $ | 1,160 | $ | 2,092 | ||||
| Foreign(1) | 666 | 1,423 | ||||||
| Total revenues | $ | 1,826 | $ | 3,515 | ||||
(1) Foreign revenues are primarily generated from grants in Israel.
Marketing
Therapeutic Products and Medical Devices
Because our planned therapeutic products and medical devices are still in the research and development stage, we will not initially need to have our own marketing personnel. If we or our subsidiaries are successful in developing marketable therapeutic products and medical devices, we will need to build our own marketing and distribution capability for those products, which would require the investment of significant financial and management resources, or we and our subsidiaries will need to find collaborative marketing partners, independent sales representatives, or wholesale distributors for the commercial sale of those products.
If we market products through arrangements with third parties, we may pay sales commissions to sales representatives or we may sell or consign products to distributors at wholesale prices. This means that our gross profit from product sales may be less than would be the case if we were to sell our products directly to end users at retail prices through our own sales force. On the other hand, selling to distributors or through independent sales representatives would allow us to avoid the cost of hiring and training our own sales employees. There can be no assurance we will be able to negotiate distribution or sales agreements with third parties on favorable terms to justify our investment in our products or achieve sufficient revenues to support our operations.
Competition
We face substantial competition in all fields of business in which we engage. That competition is likely to intensify as new products and technologies reach the market. Superior new products are likely to sell for higher prices and generate higher profit margins if acceptance by the medical community is achieved. Those companies that are successful at being the first to introduce new products and technologies to the market may gain significant economic advantages over their competitors in the establishment of a customer base and track record for the performance of their products and technologies. Such companies will also benefit from revenues from sales that could be used to strengthen their research and development, production, and marketing resources. Companies engaged in the medical products industry face the risk of obsolescence of their products and technologies as more advanced or cost-effective products and technologies are developed by competitors. As the industry matures, companies will compete based upon the performance and cost-effectiveness of their products. Specific efforts in the development of treatments for dry AMD include, but are not limited to, neuroprotection, reducing by-product accumulation, and suppressing inflammation. Specific approaches include small molecules, antibodies, and cell therapies. Some of these efforts have reached clinical development and at least one approach, complement inhibition, is currently in a Phase 3 clinical trial. We believe that replacing the entire cell rather than attempts to fix one aberrant pathway or signal confer a greater probability of success for individuals suffering with dry AMD.
| 25 |
Products for Regenerative Medicine
The cell therapy industry is characterized by rapidly evolving technology and intense competition. Our competitors include major multinational pharmaceutical companies, specialty biotechnology companies, and chemical and medical products companies operating in the fields of regenerative medicine, cell therapy, tissue engineering, and tissue regeneration. Many of these companies are well established and possess technical, research and development, financial, and sales and marketing resources significantly greater than ours. In addition, certain smaller biotech companies have formed strategic collaborations, partnerships, and other types of joint ventures with larger, well-established industry competitors that afford the smaller companies’ potential research and development as well as commercialization advantages. Academic institutions, governmental agencies, and other public and private research organizations are also conducting and financing research activities, which may produce products directly competitive to those we are developing.
We believe that some of our competitors are trying to develop pluripotent cells and human embryonic progenitor cell-based technologies and products that may compete with our stem cell products based on efficacy, safety, cost, and intellectual property positions. Ocata Therapeutics, Inc. (“Ocata”), which was acquired by a subsidiary of Astellas Pharma Inc. for approximately $379 million in 2016, and Retinal Patch Technologies Inc. have conducted clinical trials of hES cell products designed to treat dry AMD. If their products are proven to be safe and effective, they may reach the market ahead of OpRegen.
We may also face competition from companies that have filed patent applications relating to the propagation and differentiation of stem cells. Those companies include Ocata, which in 2015 had certain U.S. patents issue with claims directed to methods of producing RPE cells and isolating and purifying such cells. We may be required to seek licenses from these competitors in order to commercialize certain products proposed by us, and such licenses may not be granted.
| ITEM 1A. | RISK FACTORS |
Our business is subject to various risks, including those described below. You should consider the following risk factors, together with all of the other information included in this report, which could materially adversely affect our proposed operations, our business prospects, and financial condition, and the value of an investment in our business. There may be other factors that are not mentioned here or of which we are not presently aware that could also affect our business operations and prospects.
Risks Related to Our Business Operations and Capital Requirements
We have incurred operating losses since inception, and we do not know if or when we will attain profitability.
Our total operating losses for the fiscal years ended December 31, 2020 and 2019 were $26.4 million and $38.9 million, respectively, and we had an accumulated deficit of $294.1 million as of December 31, 2020. Since inception, we have incurred significant operating losses and have funded our operations primarily through sales of our equity securities and the equity securities of former subsidiaries, receipt of research grants, royalties on product sales, license revenues, sales of research products, and revenues from subscription fees and advertising revenue from database products of a former subsidiary. Substantially all of our losses have resulted from expenses incurred in connection with our research and development programs and from general and administrative costs associated with our operations. All of our product candidates will require substantial additional development time and resources before we would be able to apply for or receive regulatory approvals. We expect to continue to incur losses for the foreseeable future, and we anticipate these losses will increase substantially as we continue our development of, seek regulatory approval for and potentially commercialize any of our product candidates and seek to identify, assess, acquire, in-license or develop additional product candidates.
To become and remain profitable, we must succeed in developing and eventually commercializing products that generate significant revenue. This will require us to be successful in a range of challenging activities, including completing clinical trials and preclinical trials of our product candidates, obtaining regulatory approval for these product candidates and manufacturing, marketing and selling any products for which we may obtain regulatory approval. In addition, we are attempting to develop new medical products and technology. We may never succeed in these activities and, even if we do, may never generate revenues that are significant enough to achieve profitability.
| 26 |
We will continue to spend a substantial amount of our capital on research and development, but we might not succeed in developing products and technologies that are useful in medicine.
We are attempting to develop new medical products and technology. These new products and technologies might not prove to be safe and efficacious in the human medical applications for which they are being developed. Our research and development activities are costly, time consuming, and their results are uncertain. We incurred research and development expenses amounting to approximately $12.3 million and $17.9 million during the fiscal years ended December 31, 2020 and 2019, respectively. If we successfully develop a new technology or product, refinement of the new technology or product and definition of the practical applications and limitations of the technology or product may take years and require large sums of money. Clinical trials of new therapeutic products, particularly those products that are regulated as biologics, drugs, or devices, are very expensive and take years to complete. We may not have the financial resources to fund clinical trials on our own and we may have to enter into licensing or collaborative arrangements with others. Any such arrangements may be dilutive to our ownership or economic interest in the products we develop, and we might have to accept royalty payments on product sales rather than receiving the gross revenues from product sales. In addition, we may discontinue one or more of the research or product development programs. Our product and technology development programs may be delayed or discontinued should adequate funding on acceptable terms not be available.
The amount and pace of research and development work that we can do or sponsor, and our ability to commence and complete clinical trials required to obtain regulatory approval to market our therapeutic and medical device products, depends upon the amount of funds we have.
At December 31, 2020, we had $41.6 million of cash, cash equivalents and marketable equity securities. There can be no assurance that we will be able to raise additional funds on favorable terms or at all, or that any funds raised will be sufficient to permit us to develop and market our products and technology, if and when approved. Our ability to raise additional funds may be adversely impacted by deteriorating global economic conditions and the disruptions to and volatility in the credit and financial markets in the United States and worldwide resulting from the ongoing COVID-19 pandemic. Unless we are able to generate sufficient revenue or raise additional funds when needed, it is likely that we will be unable to continue our planned activities, even if we make progress in our research and development projects. We may have to postpone or limit the pace of our research and development work and planned clinical trials of our product candidates unless our cash resources increase through a growth in revenues, royalties, license fees, equity financings or borrowings.
We will need to issue additional equity or debt securities in order to raise additional capital needed to pay our operating expenses.
We expect to continue to incur substantial research and product development expenses and will need to raise additional capital to pay operating expenses until we are able to generate sufficient revenues from product sales, royalties and license fees. Our ability to raise additional equity or debt capital will depend, not only on progress made in developing new products and technologies, but also on access to capital and conditions in the capital markets. We believe that our cash, cash equivalents and marketable securities as of December 31, 2020 will be sufficient to fund our planned operations for at least the next 12 months. We have based these estimates on assumptions that may prove to be wrong, and we may use our capital resources sooner than we currently expect. Our operating plans and other demands on our cash resources may change as a result of many factors currently unknown to us, and we may need to seek additional funds sooner than planned. Any equity capital raise could result in the dilution of the interests of shareholders or may otherwise limit our ability to finance further in the future, which may negatively impact our business and operations. Any debt capital financing may involve covenants that restrict our operations, including limitations on additional borrowing and on the use of our assets. If we raise capital through licensing arrangements, it may be necessary to grant licenses on terms that are not favorable to us. There can be no assurance that we will be able to raise capital on favorable terms, or at all, or at times and in amounts needed to successfully finance product development, clinical trials, and general operations.
Lawsuits have been filed and other lawsuits may be filed against Lineage and certain members of the Lineage and Asterias Biotherapeutics, Inc. (“Asterias”) boards of directors relating to our acquisition of Asterias (the “Asterias Merger”). An adverse ruling in any such lawsuit may result in additional payments and costs.
A putative class action lawsuit alleging breach of fiduciary duties in connection with the Asterias Merger is pending in the Delaware Chancery Court. As of December 31, 2020, the defendants are certain former members of Asterias’ board of directors and Lineage. The complaint alleges that the merger process was conflicted, that the consideration was inadequate, and that the proxy statement filed by Asterias was misleading. The complaint seeks, among other things, certification of a class, rescission of the merger or monetary damages, and attorneys’ fees and costs.
| 27 |
The defendants specifically deny all allegations in the litigation and intend to defend it vigorously. However, any adverse ruling in this case could result in additional payments. Additional lawsuits arising out of or relating to the merger agreement and/or the merger may be filed in the future.
Changes in tax laws or regulations that are applied adversely to us or our customers may have a material adverse effect on our business, cash flow, financial condition or results of operations.
New income, sales, use or other tax laws, statutes, rules, regulations or ordinances could be enacted at any time, which could adversely affect our business operations and financial performance. Further, existing tax laws, statutes, rules, regulations or ordinances could be interpreted, changed, modified or applied adversely to us. For example, the 2017 Tax Act, enacted many significant changes to the U.S. tax laws. Future guidance from the Internal Revenue Service and other tax authorities with respect to the 2017 Tax Act may affect us, and certain aspects of the 2017 Tax Act could be repealed or modified in future legislation. For example, the Coronavirus Aid, Relief, and Economic Security Act (the “CARES Act”) and the Consolidated Appropriations Act, 2021 (CA) modified certain provisions of the 2017 Tax Act. In addition, it is uncertain if and to what extent various states will conform to the 2017 Tax Act, the CARES Act, or any newly enacted federal tax legislation. Changes in corporate tax rates, the realization of net deferred tax assets relating to our operations, the taxation of foreign earnings, and the deductibility of expenses under the 2017 Tax Act or future reform legislation could have a material impact on the value of our deferred tax assets, could result in significant one-time charges, and could increase our future U.S. tax expense.
Our ability to use net operating losses and other tax attributes to offset future taxable income or taxes may be subject to limitations.
As of December 31, 2020, we had net operating loss (“NOL”) carryforwards for U.S. federal and state tax purposes of approximately $169.9 million and $118.6 million, respectively. Included in these amounts are NOLs acquired through the merger with Asterias (see below). A portion of the federal and state NOL carryforwards will begin to expire, if not utilized, in varying amounts between 2027 and 2037. NOLs that expire unused will be unavailable to offset future income tax liabilities. Under federal income tax law, federal NOLs incurred in tax years beginning after December 31, 2017, may be carried forward indefinitely, but the deductibility of such NOLs in tax years beginning after December 31, 2020, is limited to 80% of taxable income. It is uncertain if and to what extent various states that we may operate in will conform to the federal tax law. In addition, under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended (the “IRC”), and corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50% change, by value, in its equity ownership over a three-year period, the corporation’s ability to use its pre-change NOL carryforwards and other pre-change tax attributes to offset its post-change income or taxes may be limited. We may experience ownership changes in the future as a result of subsequent shifts in our stock ownership, some of which may be outside of our control. If an ownership change occurs and our ability to use our NOL carryforwards is materially limited, it would harm our future operating results by effectively increasing our future tax obligations. In addition, at the state level, there may be periods during which the use of net operating loss carryforwards is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed. For example, in 2020 California enacted A.B. 85 which imposed limits on the usability of California state net operating losses and certain tax credits in tax years beginning after 2019 and before 2023.
As part of the merger with Asterias, we acquired various tax attribute carryforwards including federal and California NOLs of $52.8 million and $41.9 million, respectively, as well as California research and development credits of $2.4 million. As a result of the merger, Asterias incurred an ownership change under Section 382 of the IRC, which places annual limits on the amount of these NOLs that are available to offset income. Because of the annual limitation, the total amount of these NOLs is not immediately available to offset future income. The California research and development credit of $2.4 million has no expiration date.
| 28 |
Taxing authorities could reallocate our taxable income among our subsidiaries, which could increase our overall tax liability.
We are organized in the United States, and currently have subsidiaries in Israel and Singapore. If we succeed in growing our business, we expect to conduct increased operations through subsidiaries in various tax jurisdictions pursuant to transfer pricing arrangements between us and our subsidiaries. If two or more affiliated companies are located in different countries, the tax laws or regulations of each country generally will require that such arrangements be priced the same as those between unrelated companies dealing at arm’s length and that appropriate documentation is maintained to support the value of such arrangements. Our transfer pricing policies were formulated with the assistance of third-party experts. We are in the process of obtaining a formal transfer pricing report. However, after we receive such report, we do not intend to amend our returns for prior years. Whether we obtain a formal transfer pricing study with outside experts or not, our transfer pricing procedures will not be binding on applicable tax authorities.
If tax authorities in any of these countries were to successfully challenge our transfer prices as not reflecting arm’s length transactions, they could require us to adjust our transfer prices and thereby reallocate our income to reflect these revised transfer prices, which could result in a higher tax liability to us. In addition, if the country from which the income is reallocated does not agree with the reallocation, both countries could tax the same income, resulting in double taxation. If tax authorities were to allocate income to a higher tax jurisdiction, subject our income to double taxation or assess interest and penalties, it would increase our tax liability, which could adversely affect our financial condition, results of operations and cash flows.
Our business and operations could suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems and those of our contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters including earthquakes and tsunamis, terrorism, war, and telecommunication and electrical failures. Such events could cause significant interruption of our operations and development programs. For example, the loss of data for our product candidates could result in delays in our regulatory filings and development efforts and significantly increase our costs. To the extent that any disruption or security breach was to result in a loss of or damage to our data, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the development of our product candidates could be delayed.
In addition, our product candidates are manufactured by starting with cells that are stored in a cryopreserved master cell bank. While we believe we have adequate backup should any cell bank be lost in a catastrophic event, we or our third-party suppliers and manufacturers could lose multiple cell banks, which would severely affect our manufacturing activities. We cannot assure you that any stability or other issues relating to the manufacture of any of our product candidates or products will not occur in the future. Any delay or interruption in the supply of clinical trial supplies could delay the completion of planned clinical trials, increase the costs associated with maintaining clinical trial programs and, depending upon the period of delay, require us to commence new clinical trials at additional expense or terminate clinical trials completely. Any adverse developments affecting clinical or commercial manufacturing of our product candidates or products may result in shipment delays, inventory shortages, lot failures, product withdrawals or recalls or other interruptions in the supply of our product candidates or products. Accordingly, failures or difficulties faced at any level of our supply chain could adversely affect our business and delay or impede the development and commercialization of any of our product candidates or products and could have an adverse effect on our business, prospects, financial condition and results of operations.
Significant disruptions of information technology systems or data security breaches could adversely affect our business.
We are increasingly dependent on information technology systems and infrastructure to operate our business. In the ordinary course of our business, we collect, store, process and transmit large amounts of confidential information, including intellectual property, proprietary business information and personal information. It is critical that we do so in a secure manner to maintain the confidentiality, integrity and availability of such information. We have also outsourced some of our operations (including parts of our information technology infrastructure) to a number of third-party vendors who may have, or could gain, access to our confidential information. In addition, many of those third parties, in turn, subcontract or outsource some of their responsibilities to third parties.
| 29 |
Our information technology systems are large and complex and store large amounts of confidential information. The size and complexity of these systems make them potentially vulnerable to service interruptions or to security breaches from inadvertent or intentional actions by our employees, third party vendors and/or business partners, or from cyber-attacks by malicious third parties. Attacks of this nature are increasing in frequency, persistence, sophistication and intensity, and are being conducted by sophisticated and organized groups and individuals with a wide range of motives (including, but not limited to, industrial espionage) and expertise, including organized criminal groups, “hacktivists,” nation states and others. In addition to the extraction of important information, such attacks could include the deployment of harmful malware, ransomware, denial-of-service attacks, social engineering and other means to affect service reliability and threaten the confidentiality, integrity and availability of our information. Although the aggregate impact on our operations and financial condition has not been material to date, we have been the target of events of this nature and expect them to continue.
Significant disruptions of our, our third party vendors’ and/or business partners’ information technology systems or security breaches could adversely affect our business operations and/or result in the loss, misappropriation, and/or unauthorized access, use or disclosure of, or the prevention of access to, confidential information (including trade secrets or other intellectual property, proprietary business information and personal information), and could result in financial, legal, business and reputational harm to us. Any such event that leads to unauthorized access, use or disclosure of personal information, including personal information regarding our patients or employees, could harm our reputation, compel us to comply with federal and/or state breach notification laws and foreign law equivalents, subject us to mandatory corrective action, require us to verify the correctness of database contents and otherwise subject us to liability under laws and regulations that protect the privacy and security of personal information, which could disrupt our business, result in increased costs or loss of revenue, and/or result in significant legal and financial exposure. In addition, security breaches and other inappropriate access can be difficult to detect, and any delay in identifying them may further harm us. Moreover, the prevalent use of mobile devices to access confidential information increases the risk of security breaches. While we have implemented security measures to protect our information technology systems and infrastructure, there can be no assurance that such measures will prevent service interruptions or security breaches that could adversely affect our business. In addition, failure to maintain effective internal accounting controls related to security breaches and cybersecurity in general could impact our ability to produce timely and accurate financial statements and subject us to regulatory scrutiny.
Our business could be adversely affected if we lose the services of the key personnel upon whom we depend or if we fail to attract senior management and key scientific personnel.
We believe that our continued success depends to a significant extent upon our efforts and ability to retain highly qualified personnel, including our Chief Executive Officer, Brian Culley. All of our officers and other employees are at-will employees and may terminate their employment with us at any time with no advance notice. The loss of the services of Mr. Culley or other members of our senior management could have a material adverse effect on us. Further, the replacement of any of such individuals likely would involve significant time and costs and may significantly delay or prevent the achievement of our business and clinical objectives and would harm our business.
In addition, we could experience difficulties attracting qualified employees in the future. For example, competition for qualified personnel in the biotechnology and medical device field is intense due to the limited number of individuals who possess the skills and experience required by our industry. We will need to hire additional personnel, including experienced sales representatives, as we expand our clinical development and commercial activities. We may not be able to attract quality personnel on acceptable terms, or at all. In addition, to the extent we hire personnel from competitors, we may be subject to allegations that they have been improperly solicited or that they have divulged proprietary or other confidential information or that their former employers own their research output.
The value of our investments in public companies fluctuates based on their respective stock prices and could be negatively affected by business, regulatory and other risks applicable to them.
As of December 31, 2020, we had an equity investment in OncoCyte, a U.S. publicly traded company. As of December 31, 2020, the value of our investment in OncoCyte was approximately $8.7 million based on its closing stock price as of that date. If OncoCyte were to have delays in clinical trials or commercialization activities or otherwise realize the specific business, regulatory and other risks applicable to them, the value of its common stock and the valuation of our investment could be negatively affected. If OncoCyte were to fail and ultimately cease operations, we may lose the entire value of our investment. In addition, the value of our marketable equity securities may be significantly and adversely impacted by deteriorating global economic conditions and the disruptions to and volatility in the credit and financial markets in the United States and worldwide resulting from the ongoing COVID-19 pandemic.
| 30 |
Failure of our internal control over financial reporting could harm our business and financial results.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Because of its inherent limitations, internal control over financial reporting is not intended to provide absolute assurance that a misstatement of our financial statements would be prevented or detected. Our growth and entry into new products, technologies and markets will place significant additional pressure on our system of internal control over financial reporting. Any failure to maintain an effective system of internal control over financial reporting could limit our ability to report our financial results accurately and timely or to detect and prevent fraud. Operating our business through subsidiaries, some of which are located in foreign countries, also adds to the complexity of our internal control over financial reporting and adds to the risk of a system failure, an undetected improper use or expenditure of funds or other resources by a subsidiary, or a failure to properly report a transaction or financial results of a subsidiary. We allocate certain expenses among Lineage itself and one or more of our subsidiaries, which creates a risk that the allocations we make may not accurately reflect the benefit of an expenditure or use of financial or other resources by Lineage as the parent company and the subsidiaries among which the allocations are made. An inaccurate allocation may impact our consolidated financial results, particularly in the case of subsidiaries that we do not wholly own since our financial statements include adjustments to reflect the minority ownership interests in our subsidiaries held by others.
If we identify material weaknesses in our internal control over financial reporting, if we are unable to comply with the requirements of Section 404 of the Sarbanes-Oxley Act in a timely manner or assert that our internal control over financial reporting is effective, or if our independent registered public accounting firm is unable to express an opinion or expresses a qualified or adverse opinion about the effectiveness of our internal control over financial reporting, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our common shares could be negatively affected. In addition, we could become subject to investigations by the NYSE American, the Securities and Exchange Commission, and other regulatory authorities, which could require additional financial and management resources.
We received a loan under the Paycheck Protection Program of the CARES Act, and all or a portion of the loan may not be forgivable.
In April 2020, we received a loan for $523,305 from Axos Bank under the Paycheck Protection Program (“PPP”) contained within the new CARES Act. The PPP loan has a term of two years, is unsecured, and is guaranteed by the U.S. Small Business Administration (SBA). The loan carries a fixed interest rate of one percent per annum, with the first six months of interest deferred. Under the CARES Act and Paycheck Protection Program Flexibility Act, we are eligible to apply for forgiveness of all loan proceeds used to pay payroll costs, rent, utilities and other qualifying expenses during the 24-week period following receipt of the loan, provided that we maintain our number of employees and compensation within certain parameters during such period. Not more than 40% of the forgiven amount may be for non-payroll costs. If the conditions outlined in the PPP loan program are adhered to by us, all or part of such loan could be forgiven. However, we cannot provide any assurance that any amount of the PPP loan will ultimately be forgiven by the SBA. Any forgiven amounts will not be included in our taxable income. We applied for full forgiveness of the PPP loan on September 30, 2020.
Risks Related to Government Regulation
We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, including anti-kickback and false claims laws, transparency laws, and health information privacy and security laws. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
Our current and future operations may be subject to various federal and state fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute, the federal False Claims Act, and healthcare professional transparency laws and regulations. These laws may impact, among other things, our research activities and our proposed sales, marketing, and education programs. In addition, we may be subject to patient privacy regulation by both the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include:
| ● | the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; |
| 31 |
| ● | federal civil and criminal false claims laws, including the federal False Claims Act, and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent; | |
| ● | the federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which created new federal criminal statutes that prohibit, among other things, executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters; |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, (“HITECH”) and their implementing regulations, which imposes certain requirements on covered entities,” including certain healthcare providers, health plans, and healthcare clearinghouses, as well as their respective “business associates” that create, receive, maintain or transmit individually identifiable health information for or on behalf of a covered entity, and their subcontractors that use, disclose, access, or otherwise process individually identifiable protected health information, relating to the privacy, security, and transmission of individually identifiable health information; | |
| ● | The Physician Payments Sunshine Act which requires manufacturers of drugs, devices, biologics, and medical supplies to report annually to CMS information related to payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists, and chiropractors) and teaching hospitals, and ownership and investment interests held by physicians and other healthcare providers and their immediate family members and applicable group purchasing organizations, and, beginning in 2020 will require applicable manufacturers to report information regarding payments and other transfers of value provided during the previous year to physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists and anesthesiologist assistants, and certified nurse-midwives; and | |
| ● | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payors, including commercial insurers, state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers, marketing expenditures, or drug pricing, state and local laws that require the registration of pharmaceutical sales representatives, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. |
Because of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. In addition, recent health care reform legislation has strengthened these laws.
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply, we may be subject to significant penalties, including administrative, civil and criminal penalties, damages, fines, disgorgement, exclusion from participation in government health care programs, such as Medicare and Medicaid, integrity oversight and reporting obligations, imprisonment, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
| 32 |
If we do not receive regulatory approvals, we will not be permitted to sell our therapeutic and medical device products.
The therapeutic and medical device products that we and our subsidiaries develop cannot be sold until the FDA and corresponding foreign regulatory authorities approve the products for medical use. The need to obtain regulatory approval to market a new product means that:
| ● | We will have to conduct expensive and time-consuming clinical trials of new products. The full cost of conducting and completing clinical trials necessary to obtain FDA and foreign regulatory approval of a new product cannot be presently determined but could exceed our current financial resources. | |
| ● | Clinical trials and the regulatory approval process for a pharmaceutical or cell-based product can take several years to complete. As a result, we will incur the expense and delay inherent in seeking FDA and foreign regulatory approval of new products, even if the results of clinical trials are favorable. | |
| ● | Data obtained from preclinical and clinical studies is susceptible to varying interpretations and regulatory changes that could delay, limit, or prevent regulatory agency approvals. | |
| ● | Because the therapeutic products we are developing with pluripotent stem cell technology involve the application of new technologies and approaches to medicine, the FDA or foreign regulatory agencies may subject those products to additional or more stringent review than drugs or biologics derived from other technologies. | |
| ● | A product that is approved may be subject to restrictions on use. | |
| ● | The FDA can recall or withdraw approval of a product, if it deems necessary. | |
| ● | We will face similar regulatory issues in foreign countries. |
Government-imposed bans or restrictions and religious, moral, and ethical concerns about the use of hES cells could prevent us from developing and successfully marketing stem cell products.
Government-imposed bans or restrictions on the use of embryos or hES cells in research and development in the United States and abroad could generally constrain stem cell research, thereby limiting the market and demand for our products. During March 2009, President Obama lifted certain restrictions on federal funding of research involving the use of hES cells, and in accordance with President Obama’s Executive Order, the National Institutes of Health (“NIH”) has adopted guidelines for determining the eligibility of hES cell lines for use in federally funded research. The central focus of the guidelines is to assure that hES cells used in federally funded research were derived from human embryos that were created for reproductive purposes, were no longer needed for this purpose, and were voluntarily donated for research purposes with the informed written consent of the donors. The hES cells that were derived from embryos created for research purposes rather than reproductive purposes, and other hES cells that were not derived in compliance with the guidelines, are not eligible for use in federally funded research. California law requires that stem cell research be conducted under the oversight of a stem cell review oversight committee (“SCRO”). Many kinds of stem cell research, including the derivation of new hES cell lines, may only be conducted in California with the prior written approval of the SCRO. A SCRO could prohibit or impose restrictions on the research that we plan to do. The use of hES cells may give rise to religious, moral, and ethical issues. These considerations could lead to more restrictive government regulations or could generally constrain stem cell research, thereby limiting the market and demand for our products.
We expect that the commercial opportunity for some of our products may depend on our ability to obtain reimbursement and continued coverage from various payors, including government entities and insurance companies.
If these third-party payors do not consider our products to be cost-effective compared to other therapies, they may not cover our products as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
| 33 |
For example, in the United States, healthcare providers are reimbursed for covered services and products they deliver through Medicare, Medicaid and other government healthcare programs, as well as through private payers. No uniform policy for coverage and reimbursement exists in the United States, and coverage and reimbursement can differ significantly from payor to payor. Decisions regarding whether to cover any of our product candidates, if approved, the extent of coverage and amount of reimbursement to be provided are made on a plan-by-plan basis. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement rates, but also have their own methods and approval process apart from Medicare determinations. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our product candidates to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. We may be required to provide specified rebates or discounts on the products we sell to certain government funded programs, including Medicare and Medicaid, and those rebates or discounts have increased over time. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, the “ACA”), enacted in 2010, increased many of the mandatory discounts and rebates and imposed a new branded prescription pharmaceutical manufacturers and importers fee payable each year by certain manufacturers.
We face similar issues outside of the United States. In some non-U.S. jurisdictions, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the EU provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product, or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, products launched in the EU do not follow price structures of the United States and generally tend to be significantly lower.
Disruptions at the FDA and other government agencies caused by funding shortages or global health concerns could negatively impact our business.
The ability of the FDA to review and approve proposed clinical trials or new product candidates can be affected by a variety of factors, including, but not limited to, government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, statutory, regulatory, and policy changes, and other events that may otherwise affect the FDA’s ability to perform routine functions. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new product candidates to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, including for 35 days beginning on December 22, 2018, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA, have had to furlough critical FDA employees and stop critical activities.
Separately, in response to the global COVID-19 pandemic, in March 2020, the FDA announced its intention to postpone most foreign inspections of manufacturing facilities and temporarily postponed routine surveillance inspections of domestic manufacturing facilities. In July 2020 domestic inspections restarted only on a risk-based basis. Regulatory authorities outside the United States may adopt similar restrictions or other policy measures in response to the COVID-19 pandemic. If a prolonged government shutdown occurs, or if global health concerns continue to prevent the FDA or other regulatory authorities from conducting their regular inspections, reviews, or other regulatory activities, it could significantly impact the ability of the FDA or other regulatory authorities to timely review and process our regulatory submissions, which could have a material adverse effect on our business.
The ACA and future changes to that law may adversely affect our business.
As a result of the adoption of the ACA, in the United States, substantial changes have been made to the system for paying for healthcare in the United States. Among the ACA’s provisions of importance to our industry are that it:
| ● | created the branded prescription pharmaceutical manufacturers and importers annual fee; | |
| ● | increased the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program, to 23.1% and 13% of the average manufacturer price for most branded and generic drugs, respectively and capped the total rebate amount for innovator drugs at 100% of the Average Manufacturer Price; |
| 34 |
| ● | created new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for certain drugs and biologics that are inhaled, infused, instilled, implanted or injected; | |
| ● | extended manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| ● | expanded eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for individuals with income at or below 133% of the federal poverty level, thereby potentially increasing manufacturers’ Medicaid rebate liability; | |
| ● | expanded the entities eligible for discounts under the Public Health program; | |
| ● | created a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; | |
| ● | established a Center for Medicare & Medicaid Innovation at the Centers for Medicare & Medicaid Services (“CMS”) to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending; and | |
| ● | created a licensure framework for follow on biologic products. |
There remain judicial and Congressional challenges to certain aspects of the ACA, as well as efforts by the Trump administration to repeal or replace certain aspects of the ACA. Since January 2017, President Trump signed Executive Orders and other directives designed to delay the implementation of certain provisions of the ACA. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the ACA. While Congress has not passed comprehensive repeal legislation, it has enacted laws that modify certain provisions of the ACA such as removing penalties, starting January 1, 2019, for not complying with the ACA’s individual mandate to carry health insurance, and eliminating the implementation of certain ACA-mandated fees. On December 14, 2018, a Texas U.S. District Court Judge ruled that the ACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress as part of the 2017 Tax Act. Additionally, on December 18, 2019, the U.S. Court of Appeals for the 5th Circuit upheld the District Court ruling that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. The U.S. Supreme Court is currently reviewing the case, although it is uncertain when or how the Supreme Court will rule. It is unclear how such litigation and other efforts to repeal and replace the ACA will impact the ACA and our business.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. For example, the Budget Control Act of 2011, includes reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute, will remain in effect through 2030, with the exception of a temporary suspension from May 1, 2020 through March 31, 2021, unless additional Congressional action is taken. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, reduced Medicare payments to several providers, including hospitals, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
| 35 |
Further, there has been heightened governmental scrutiny in the United States of pharmaceutical pricing practices in light of the rising cost of prescription drugs and biologics. Such scrutiny has resulted in several recent congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for products. At the federal level, the Trump administration’s budget proposal for fiscal year 2021 includes a $135 billion allowance to support legislative proposals seeking to reduce drug prices, increase competition, lower out-of-pocket drug costs for patients, and increase patient access to lower-cost generic and biosimilar drugs. On March 10, 2020, the Trump administration sent “principles” for drug pricing to Congress, calling for legislation that would, among other things, cap Medicare Part D beneficiary out-of-pocket pharmacy expenses, provide an option to cap Medicare Part D beneficiary monthly out-of-pocket expenses, and place limits on pharmaceutical price increases. In addition, the Trump administration previously released a “Blueprint” to lower drug prices and reduce out of pocket costs of drugs that contained proposals to increase drug manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products, and reduce the out-of-pocket costs of drug products paid by consumers. HHS has solicited feedback on some of these measures and has implemented others under its existing authority. For example, in May 2019, CMS issued a final rule to allow Medicare Advantage plans the option to use step therapy for Part B drugs beginning January 1, 2020. This final rule codified CMS’s policy change that was effective January 1, 2019. Further, on November 20, 2020, HHS finalized a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers. The likelihood of implementation of any of the other Trump administration reform initiatives is uncertain, particularly in light of the new presidential administration. On November 20, 2020, CMS issued an interim final rule implementing President Trump’s Most Favored Nation executive order, which would tie Medicare Part B payments for certain physician-administered drugs to the lowest price paid in other economically advanced countries, effective January 1, 2021. On December 28, 2020, the United States District Court in Northern California issued a nationwide preliminary injunction against implementation of the interim final rule. The likelihood of implementation of any of the other Trump administration reform initiatives is uncertain, particularly in light of the new presidential administration. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
In addition, it is possible that additional governmental action is taken to address the COVID-19 pandemic.
If we fail to comply with the extensive legal and regulatory requirements affecting the health care industry, we could face increased costs, penalties and a loss of business.
Our activities, and the activities of our collaborators, distributors and other third-party providers, are subject to extensive government regulation and oversight both in the U.S. and in foreign jurisdictions. The FDA and comparable agencies in other jurisdictions will directly regulate many of our most critical business activities, including the conduct of preclinical and clinical studies, product manufacturing, advertising and promotion, product distribution, adverse event reporting and product risk management. Our interactions in the U.S. or abroad with physicians and other health care providers that may prescribe or purchase our products are also subject to government regulation designed to prevent fraud and abuse in the sale and use of the products and place greater restrictions on the marketing practices of health care companies. Health care companies are facing heightened scrutiny of their relationships with health care providers from anti-corruption enforcement officials. In addition, health care companies have been the target of lawsuits and investigations alleging violations of government regulation, including claims asserting submission of incorrect pricing information, impermissible off-label promotion of pharmaceutical products, payments intended to influence the referral of health care business, submission of false claims for government reimbursement, antitrust violations or violations related to environmental matters. Risks relating to compliance with laws and regulations may be heightened as we bring products to the market globally.
Regulations governing the health care industry are subject to change, with possibly retroactive effect, including:
| ● | new laws, regulations or judicial decisions, or new interpretations of existing laws, regulations or decisions, related to health care availability, pricing or marketing practices, compliance with wage and hour laws and other employment practices, method of delivery, payment for health care products and services, compliance with health information and data privacy and security laws and regulations, tracking and reporting payments and other transfers of value made to physicians and teaching hospitals, extensive anti-bribery and anti-corruption prohibitions, product serialization and labeling requirements and used product take-back requirements; | |
| ● | changes in the FDA and foreign regulatory approval processes that may delay or prevent the approval of new products and result in lost market opportunity; |
| 36 |
| ● | requirements that provide for increased transparency of clinical trial results and quality data, such as the EMA’s clinical transparency policy, which could impact our ability to protect trade secrets and competitively sensitive information contained in approval applications or could be misinterpreted leading to reputational damage, misperception or legal action which could harm our business; and | |
| ● | changes in FDA and foreign regulations that may require additional safety monitoring, labeling changes, restrictions on product distribution or use, or other measures after the introduction of our products to market, which could increase our costs of doing business, adversely affect the future permitted uses of approved products, or otherwise adversely affect the market for our products. |
Violations of governmental regulation may be punishable by criminal and civil sanctions against us, including fines and civil monetary penalties and exclusion from participation in government programs, including Medicare and Medicaid, as well as against executives overseeing our business. In addition to penalties for violation of laws and regulations, we could be required to repay amounts we received from government payors or pay additional rebates and interest if we are found to have miscalculated the pricing information we have submitted to the government. We cannot ensure that our compliance controls, policies and procedures will in every instance protect us from acts committed by our employees, collaborators, partners or third-party providers that would violate the laws or regulations of the jurisdictions in which we operate. Whether or not we have complied with the law, an investigation into alleged unlawful conduct could increase our expenses, damage our reputation, divert management time and attention and adversely affect our business.
Even if we receive approval for our products, we may be subject to extensive regulatory obligations in order to commercialize our products.
Even after initial FDA or foreign regulatory agency approval has been obtained, further studies may be required to provide additional data on safety or to gain approval for the use of a product as a treatment for clinical indications other than those initially targeted. Use of a product during testing and after marketing could reveal side effects that could delay, impede, or prevent marketing approval, result in a regulatory agency-ordered product recall, or in regulatory agency-imposed limitations on permissible uses or in withdrawal of approval. For example, if the FDA or foreign regulatory agency becomes aware of new safety information after approval of a product, it may require us to conduct further clinical trials to assess a known or potential serious risk and to assure that the benefit of the product outweigh the risks. If we are required to conduct such a post-approval study, periodic status reports must be submitted to the FDA or foreign regulatory agency. Failure to conduct such post-approval studies in a timely manner may result in substantial civil or criminal penalties. Data resulting from these clinical trials may result in expansions or restrictions to the labeled indications for which a product has already been approved. Any of these requirements or actions may negatively impact our business or operations.
If we are deemed to be an investment company, we may have to institute burdensome compliance requirements and our activities may be restricted.
An entity that, among other things, is or holds itself out as being engaged primarily, or proposes to engage primarily, in the business of investing, reinvesting, owning, trading or holding certain types of securities would be deemed an investment company under the Investment Company Act of 1940, as amended (the “1940 Act”). Based on the securities we hold, including our equity ownership in publicly traded companies, we may not meet the requirements for an exemption promulgated under the 1940 Act. If we are deemed to be an investment company under the 1940 Act, we would be subject to additional limitations on operating our business, including limitations on the issuance of securities, which may make it difficult for us to raise capital.
Risks Related to Our Clinical Development and Commercial Operations
Clinical studies are costly, time consuming and are subject to risks that could delay or prevent commercialization of our current or future product candidates.
We cannot guarantee that any clinical studies will be conducted as planned or completed on schedule, if at all. A failure of one or more clinical studies can occur at any stage of development. Events that may prevent successful or timely completion of clinical development include but are not limited to:
| ● | inability to generate satisfactory preclinical, toxicology, or other in vivo or in vitro data or diagnostics to support the initiation or continuation of clinical studies necessary for product approval; |
| 37 |
| ● | delays in securing clinical investigators and agreeing on acceptable terms with contract research organizations (“CROs”) and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among CROs and clinical trial sites; | |
| ● | delays in obtaining required Institutional Review Board (“IRB”) approval at each clinical trial site; | |
| ● | failure to obtain permission from regulatory authorities to conduct a clinical trial after review of an investigational new drug (“IND”) or equivalent foreign application or amendment; | |
| ● | slower than anticipated rates of patient recruitment and enrollment (including as a result of actual or threatened public health emergencies and outbreaks of disease such as the current COVID-19 pandemic), failing to reach the targeted number of patients due to competition for patients from other trials, or patients dropping out of our clinical studies once enrolled; | |
| ● | failure by clinical sites or our CROs or other third parties to adhere to clinical trial requirements or report complete findings; | |
| ● | failure to perform the clinical studies in accordance with the FDA’s good clinical practices requirements or applicable foreign regulatory guidelines; | |
| ● | occurrence of adverse events associated with our product candidates or with product candidates of third parties that may have characteristics similar to or perceived to be similar to our product candidates; | |
| ● | negative or inconclusive results from our clinical trials which may result in our deciding, or regulators requiring us, to conduct additional clinical studies or to curtail or abandon development programs for a product candidate; | |
| ● | unforeseen side effects, possibly resulting in the FDA or other regulatory authorities denying approval of our product candidates; | |
| ● | approval and introduction of new therapies or changes in standards of practice or regulatory guidance that render our clinical trial endpoints or the targeting of our proposed indications obsolete; | |
| ● | inability to monitor patients adequately during or after treatment or problems with investigator or patient compliance with the trial protocols; | |
| ● | inability or unwillingness of medical investigators to follow our clinical protocols; | |
| ● | unavailability of clinical trial supplies; | |
| ● | inability to use clinical trial results from foreign jurisdictions to support U.S. regulatory approval; | |
| ● | changes in regulatory requirements and guidance that require amending or submitting new clinical protocols; | |
| ● | the cost of clinical studies of our product candidates; and | |
| ● | delays in agreeing on acceptable terms with third-party manufacturers and the time for manufacture of sufficient quantities of our product candidates for use in clinical studies. |
Any inability to successfully complete clinical development and obtain regulatory approval could result in additional costs to us or impair our ability to generate revenue. Clinical trial delays could also shorten any periods during which our products have patent protection and may allow competitors to develop and bring products to market before we do and may harm our business and results of operations.
| 38 |
Clinical and preclinical drug development involves a lengthy and expensive process with an uncertain outcome. The results of early preclinical trials and clinical trials of our product candidates are not necessarily predictive of future results. Our product candidates may not have favorable results in later clinical trials, if any, or receive regulatory approval on a timely basis, if at all.
Clinical and preclinical drug development is expensive and can take many years to complete, and its outcome is inherently uncertain. Our clinical trials may not be conducted as planned or completed on schedule, if at all, and failure can occur at any time during the preclinical trial or clinical trial process. All of our product candidates will require substantial additional development, and no assurances can be given that the development of any of our product candidates will ultimately be successful. Although we may from time to time disclose results from preclinical testing or preliminary data or interim results from our clinical studies of our product candidates, and earlier clinical studies, including clinical studies with similar product candidates, these are not necessarily predictive of future results, including clinical trial results. The historical failure rate for product candidates in our industry is high.
The results of our current and future clinical trials may differ from results achieved in earlier preclinical and clinical studies for a variety of reasons, including:
| ● | we may not demonstrate the potency and efficacy benefits observed in previous studies; | |
| ● | our efforts to improve, standardize and automate the manufacture of our product candidates, including OpRegen, OPC1 and VAC2, and any resulting deviations in the manufacture of our product candidates, may adversely affect the safety, purity, potency or efficacy of such product candidates; | |
| ● | differences in trial design, including differences in size, eligibility criteria, and patient populations; | |
| ● | advancements in the standard of care may affect our ability to demonstrate efficacy or achieve trial endpoints in our current or future clinical trials; | |
| ● | safety issues or adverse events in patients that enroll in our current or future clinical trials; and | |
| ● | results in preclinical and clinical tests may not be repeated in subsequent tests or be predictive of future results. |
In particular, data presented from the Phase 1/2a open-label trial showed that both the surgical procedure and the OpRegen cells were generally well tolerated, with no treatment-related systemic serious adverse events reported to date in the first nine patients. The best corrected visual acuity of these patients remained relatively stable. In addition, the imaging of patients 8 and 9 suggested early signs of structural improvement within the retina. However, we do not know how OpRegen will perform in future clinical trials.
It is not uncommon to observe results in clinical trials that are unexpected based on preclinical trials and early clinical trials, and many product candidates fail in clinical trials despite very promising early results. Moreover, preclinical and clinical data may be susceptible to varying interpretations and analyses. A number of companies in the biotechnology industry have suffered significant setbacks in clinical development even after achieving promising results in earlier studies.
Further, as a result of the COVID-19 pandemic, if patients drop out of our clinical trials, miss scheduled doses or follow-up visits or otherwise fail to follow clinical trial protocols, or if our clinical trials are otherwise disrupted due to COVID-19 or actions taken to slow its spread, the integrity of data from our clinical trials may be compromised or not accepted by the FDA or other regulatory authorities, which would represent a significant setback for the applicable program.
Even if our current and planned clinical trials are successful, we will need to conduct additional clinical trials, which may include registrational trials, trials in additional patient populations or under different treatment conditions, and trials using different manufacturing protocols, processes, materials or facilities or under different manufacturing conditions, before we are able to seek approvals for our product candidates from the FDA and regulatory authorities outside the United States to market and sell these product candidates. Our failure to meet the requirements to support marketing approval for our product candidates in our ongoing and future clinical trials would substantially harm our business and prospects. For the foregoing reasons, our ongoing and planned clinical trials may not be successful, which could have a material adverse effect on our business, financial condition and results of operations.
| 39 |
Interim, topline and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose preliminary or topline data from our clinical trials, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the topline results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, topline data should be viewed with caution until the final data are available. From time to time, we may also disclose interim data from our clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Adverse differences between preliminary or interim data and final data could significantly harm our business prospects.
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our company in general. In addition, the information we choose to publicly disclose regarding a particular trial is based on what is typically extensive information, and you or others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure, and any information we determine not to disclose may ultimately be deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding a particular product candidate or our business. If the topline data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize, our product candidates may be harmed, which could harm our business, operating results, prospects or financial condition.
Because we have multiple cell therapy programs in clinical development, we may expend our limited resources to pursue a particular product candidate and fail to capitalize on product candidates that may be more profitable or for which there is a greater likelihood of success.
We have three cell therapy programs in clinical development. OpRegen is currently in a Phase 1/2a multicenter clinical trial for the treatment of dry AMD, OPC-1 is currently in a Phase 1/2a clinical trial for acute spinal cord injuries, and VAC2 is in a Phase 1 clinical trial in non-small cell lung cancer. As a result of these and other future clinical trials for these product candidates or any of our future product candidates may make our decision as to which product candidates to focus on more difficult and we may forgo or delay pursuit of opportunities with other product candidates that could have had greater commercial potential or likelihood of success.
Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and product candidates may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through future collaborations, licenses and other similar arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate.
Additionally, we may pursue additional in-licenses or acquisitions of development-stage assets or programs, which entails additional risk to us. Identifying, selecting and acquiring promising product candidates requires substantial technical, financial and human resources expertise. Efforts to do so may not result in the actual acquisition or license of a particular product candidate, potentially resulting in a diversion of our management’s time and the expenditure of our resources with no resulting benefit. For example, if we are unable to identify programs that ultimately result in approved products, we may spend material amounts of our capital and other resources evaluating, acquiring and developing products that ultimately do not provide a return on our investment.
| 40 |
The commercial success of any of our current or future product candidates will depend upon the degree of market acceptance by physicians, patients, third-party payors, other health care providers and others in the medical community.
Even if a product candidate obtains regulatory approval, its commercial success will depend in part on physicians, patients, third-party payors, other health care providers and others in the medical community accepting our product candidates as medically useful, cost-effective, and safe. Any product we bring to the market may not gain market acceptance by such parties. The degree of market acceptance of any of our products will depend on several factors, including without limitation:
| ● | the efficacy of the product as demonstrated in clinical trials and potential advantages over competing treatments; | |
| ● | the prevalence and severity of the disease and any side effects; | |
| ● | the clinical indications for which approval is granted, including any limitations or warnings contained in a product’s approved labeling; |
| ● | the convenience and ease of administration; | |
| ● | the cost of treatment, particularly as additive to existing treatments; | |
| ● | the willingness of the patients and physicians to accept and use these therapies; | |
| ● | the marketing, sales and distribution support for the products; | |
| ● | the publicity concerning our products or competing products and treatments; and | |
| ● | the pricing and availability of coverage and adequate reimbursement by third-party payors and government authorities. |
Even if a product displays a favorable efficacy and safety profile upon approval, market acceptance of the product will be uncertain. Efforts to educate the medical community and third-party payors on the benefits of the products may require significant investment and resources and may never succeed. If our products fail to achieve an adequate level of acceptance by physicians, patients, third-party payors, other health care providers and others in the medical community, we will not be able to generate sufficient revenue to become or remain profitable.
If the market opportunities for our product candidates are smaller than we believe and estimate they are, we may not meet our revenue expectations and our business may suffer.
Our projections of the number of potential users in the markets we are attempting to address are based on our beliefs and estimates. Our estimates have been derived from a variety of sources, including market research and publications and scientific literature estimating the total number of potential patients and currently approved or used therapies. Our estimates are also based on assumptions regarding the potential size of the market assuming broad regulatory approval or potential usage by physicians beyond the approved label. Any of our estimates may prove to be incorrect. The scope of approval and potential use of any product candidate may be significantly narrower, and the number of patients may turn out to be lower than expected. Competitive products or approaches may be approved or come into use and the potentially addressable patient population for each of our product candidates may be limited or may not be amenable to treatment with our product candidates, and new patients may become increasingly difficult to identify or gain access to, any which could adversely affect our results of operations and our business.
Sales of the products we may develop will be adversely affected by the availability of competing products.
Our products and product candidates will face substantial competition, whether through the development of safer and more effective alternatives to our products, lower costs to administer than our products or other forms of competition such as more favorable distribution, reimbursement and pricing or formulary and health care provider acceptance.
| 41 |
The cell therapy industry is characterized by rapidly evolving technology and intense competition. Our competitors include major multinational pharmaceutical companies, specialty biotechnology companies, and chemical and medical products companies operating in the fields of regenerative medicine, cell therapy, tissue engineering, and tissue regeneration. Many of these companies are well established and possess technical, research and development, financial, and sales and marketing resources significantly greater than ours. In addition, certain smaller biotechnology companies have formed strategic collaborations, partnerships, and other types of joint ventures with larger, well-established industry competitors that afford the smaller companies’ potential research and development as well as commercialization advantages. Academic institutions, governmental agencies, and other public and private research organizations are also conducting and financing research activities, which may produce products directly competitive to those we are developing.
We believe that some of our competitors are trying to develop pluripotent cells and human embryonic progenitor cell (“hEPC”) based technologies and products that may compete with our stem cell products based on efficacy, safety, cost, and intellectual property positions. Ocata, which was acquired by a subsidiary of Astellas Pharma Inc., and Retinal Patch Technologies Inc. are conducting clinical trials of hES cell products designed to treat age-related macular degeneration. If their products are proven to be safe and effective, they may reach the market ahead of OpRegen.
We may also face competition from companies that have filed patent applications relating to the propagation and differentiation of stem cells. Those companies include Ocata, which in 2015 had certain U.S. patents issue with claims directed to methods of producing RPE cells and isolating and purifying such cells. We may be required to seek licenses from these competitors in order to commercialize certain products proposed by us, and such licenses may not be granted.
Competitive products may make any products we develop obsolete or noncompetitive before we recover the expense of developing and commercializing our product candidates. If we are unable to compete effectively, our opportunity to generate revenue from the sale of our products we may develop, if approved, could be adversely affected.
We will face risks related to our own manufacturing capabilities and those related to our reliance on third parties to manufacture products, including those related to product acquisition costs, production delays, and supply shortages that could impair our ability to complete the development and commercialization of our product candidates.
The manufacture of medical products is complex and requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Although we have manufacturing capability through Cell Cure for OpRegen, OPC1, and VAC2 in Israel, we will need greater manufacturing capacity if we are to successfully commercialize our products. Unless we can raise the capital required to construct our own commercial scale manufacturing facilities and can develop the expertise to manage and operate a manufacturing facility of our own, we may need to rely on third-party manufacturers to manufacture any products we develop. There is no assurance that we will be able to identify manufacturers on acceptable terms or at all. Regardless of whether we do our own manufacturing or rely on third parties to manufacture products for us, we will face risks related to the manufacture of our products including these risks:
| ● | We or any third-party manufacturers might not timely formulate and manufacture our products or produce the quantity and quality required to meet our clinical and commercial needs, if any. | |
| ● | We or any third-party manufacturers may not execute our manufacturing procedures appropriately. | |
| ● | Any third-party manufacturers we engage may not perform as agreed or may not remain in the contract manufacturing business for the time required to supply our clinical trials or to successfully produce, store and distribute our products on a commercial scale. | |
| ● | We or any third-party manufacturers will be subject to ongoing periodic unannounced inspection by the FDA and corresponding state agencies to ensure strict compliance with current good manufacturing practices (“cGMP”), and other government regulations and corresponding foreign standards. We will not have control over third-party manufacturers’ compliance with applicable regulations and standards. |
| 42 |
| ● | We may not own, or may have to share, the intellectual property rights to any improvements made by our third-party manufacturers in the manufacturing process for our product candidates. | |
| ● | We may not obtain licenses for third-party intellectual property rights needed by manufacturers to produce our products. | |
| ● | Third-party manufacturers could breach or terminate their agreements with us. | |
| ● | We or third-party manufacturers may experience manufacturing difficulties as a result of resource constraints, labor disputes, unstable political environments, natural disasters, public health crises such as pandemics and epidemics, political crises such as terrorism, war, political insecurity or other conflict, or other events outside of our or our third-party manufacturers control (including as a result of actual or threatened public health emergencies and outbreaks of disease such as the current COVID-19 pandemic). This may result in business closures that affect us and our third-party manufacturers. |
In addition, we may rely on third parties to perform release testing on our product candidates prior to delivery to patients. If these tests are not appropriately conducted and test data are not reliable, patients could be put at risk of serious harm which could result in product liability suits.
If we or any third-party manufacturers we may engage were to encounter any of these difficulties, our ability to provide our product candidates to patients in clinical trials or to the medical market place would be jeopardized. Any delay or interruption in the supply of clinical trial supplies could delay the completion of clinical trials, increase the costs associated with maintaining clinical trial programs and, depending upon the period of delay, could require us to either commence new clinical trials at additional expense or terminate clinical trials completely. Each risk could delay our clinical trials, any approval of our product candidates by the FDA, or the commercialization of our product candidates, and could result in higher costs or deprive us of potential product revenue.
Any cell-based products that receive regulatory approval may be difficult and expensive to manufacture profitably.
Cell-based products are among the more expensive biologic products to manufacture in accordance with cGMP. We do not yet have sufficient information to reliably estimate the cost of commercially manufacturing any of our product candidates. Excessive manufacturing costs could make our product candidates too expensive to compete in the medical market place with alternative products manufactured by our competitors or might result in third party payors such as health insurers and Medicare, declining to cover our products or setting reimbursement levels too low for us to earn a profit from the commercialization of one or more of our products.
We may not secure a commercialization partner for Renevia.
In September 2019, Renevia was granted a CE Mark and Class III classification with an intended use in adults as a resorbable matrix for the delivery of autologous adipose tissue preparations to restore and/or augment facial volume after subcutaneous fat volume loss for the treatment of facial lipoatrophy. The CE Mark provides us, or our authorized agent, the authority to market and distribute Renevia throughout the European Union (“EU”) and in other countries that recognize the CE Mark.
However, because we have no commercial infrastructure, we are seeking a commercialization partner in the EU. We can give no assurance that we will secure a commercialization partner for Renevia or otherwise commercialize Renevia.
The ongoing COVID-19 pandemic may adversely affect our operations, including the conduct of our clinical trials.
In December 2019, a novel strain of coronavirus and the resulting illness known as COVID-19 emerged in Wuhan, China. The outbreak has now spread to other countries and has been declared a pandemic by the World Health Organization.
| 43 |
The COVID-19 pandemic has resulted in travel and other restrictions in order to reduce the spread of the disease, including a California executive order and several other state and local orders across the country, which, among other things, direct individuals to shelter at their places of residence, direct businesses and governmental agencies to cease non-essential operations at physical locations, prohibit certain non-essential gatherings, and order cessation of non-essential travel. In response to these public health directives and orders, we have implemented work-from-home policies for our employees. The effects of the executive order, the shelter-in-place order and our work-from-home policies may negatively impact productivity, disrupt our business and delay our clinical programs and timelines, the magnitude of which will depend, in part, on the length and severity of the restrictions and other limitations on our ability to conduct our business in the ordinary course. These and similar, and perhaps more severe, disruptions in our operations could negatively impact our business, operating results and financial condition.
As COVID-19 continues to spread in the United States and Israel, we have experienced and may continue to experience disruptions that could adversely affect our operations and clinical trials, including:
| ● | delays or difficulties in enrolling, or conducting follow-up visits with, patients in our clinical trials, particularly patients for our OpRegen Phase 1/2a clinical trial, who are older and who may be at higher risk of complications from COVID-19; | |
| ● | delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and staff; | |
| ● | diversion of healthcare resources away from the conduct of clinical trials; | |
| ● | interruption of key clinical trial activities, such as clinical trial site monitoring, due to limitations on travel; | |
| ● | limited availability of our employees and the staff of our current clinical sites due to sickness or social distancing measures; | |
| ● | manufacturing difficulties for us and our suppliers of raw materials caused by business closures; | |
| ● | delays in clinical sites receiving the supplies and materials needed to conduct our clinical trials, including interruption in global shipping that may affect the transport of clinical trial materials; | |
| ● | changes in local regulations as part of a response to the COVID-19 outbreak which may require us to change the ways in which our clinical trials are conducted, which may result in unexpected costs, or to discontinue the clinical trials altogether; | |
| ● | interruption or delays in the operations of the FDA or other regulatory authorities, which may impact review and approval timelines; | |
| ● | risk that participants enrolled in our clinical trials will acquire COVID-19 while the clinical trial is ongoing, which could impact the results of the clinical trial, including by increasing the number of observed adverse events; and; | |
| ● | refusal of the FDA to accept data from clinical trials in affected geographies; |
These and other disruptions in our operations and the global economy could negatively impact our business, operating results and financial condition. The extent to which the COVID-19 pandemic affects our operations will depend on future developments, which are highly uncertain and cannot be predicted with confidence, including the duration and severity of the pandemic, and the actions that may be required to contain the COVID-19 pandemic or treat its impact.
Our clinical trials have been, and may in the future be, affected by the COVID-19 pandemic. For example, the COVID-19 pandemic has impacted patient enrollment in our OpRegen Phase 1/2a multicenter clinical trial and the VAC2 Phase 1 multicenter clinical trial. In particular, some sites have paused enrollment to focus on, and direct resources to, the COVID-19 pandemic, while at other sites, patients are choosing not to enroll or continue participating in the clinical trial as a result of the pandemic. We are unable to predict with confidence the duration of such patient enrollment delays and difficulties. If patient enrollment is delayed for an extended period of time, such clinical trials could be delayed or otherwise adversely affected. Our inability to enroll a sufficient number of patients for any of our current or future clinical trials could result in significant delays or may require us to abandon one or more clinical trials altogether. As a result, we may experience new or additional delays and difficulties in enrollment, which would result in the delay of completion of such trials beyond our expected timelines.
| 44 |
Our ongoing or planned clinical trials may also be impacted by interruptions or delays in the operations of the FDA and comparable foreign regulatory agencies.
In addition, quarantines, shelter-in-place and similar government orders, or the perception that such orders, shutdowns or other restrictions on the conduct of business operations could occur, related to COVID-19 or other infectious diseases could impact personnel at our CROs or third-party manufacturing facilities upon which we rely, or the availability or cost of materials, which could disrupt the supply chain for our product candidates. To the extent our suppliers and service providers are unable to comply with their obligations under our agreements with them or they are otherwise unable to deliver or are delayed in delivering goods and services to us due to the COVID-19 pandemic, our ability to continue meeting clinical supply demand for our product candidates or otherwise advancing development of our product candidates may become impaired.
The spread of COVID-19 and actions taken to reduce its spread may also materially affect us economically. While the potential economic impact brought by, and the duration of, the COVID-19 pandemic may be difficult to assess or predict, there could be a significant disruption of global financial markets, reducing our ability to access capital, which could in the future negatively affect our liquidity and financial position. In addition, the trading prices for other biotechnology companies have been highly volatile as a result of the COVID-19 pandemic. As a result, we may face difficulties raising capital through sales of our common shares or such sales may be on unfavorable terms.
COVID-19 and actions taken to reduce its spread continue to rapidly evolve. The extent to which COVID-19 may impede the development of our product candidates, reduce the productivity of our employees, disrupt our supply chains, delay our clinical trials, reduce our access to capital or limit our business development activities, will depend on future developments, which are highly uncertain and cannot be predicted with confidence.
In addition, to the extent the ongoing COVID-19 pandemic adversely affects our business and results of operations, it may also have the effect of heightening many of the other risks and uncertainties described in this ‘‘Risk Factors’’ section.
The withdrawal of the United Kingdom (the “U.K.”) from the EU, commonly referred to as “Brexit,” may adversely impact our ability to obtain regulatory approvals of our product candidates in the EU, result in restrictions or imposition of taxes and duties for importing our product candidates into the EU, and may require us to incur additional expenses in order to develop, manufacture and commercialize our product candidates in the EU.
On June 23, 2016, the U.K. held a referendum in which a majority of the eligible members of the electorate voted for the U.K. to leave the EU. The U.K. formally left the EU on January 31, 2020, which is commonly referred to as Brexit, with a transition period that ended December 31, 2020.
| 45 |
Since a significant proportion of the regulatory framework in the U.K. applicable to our business and our product candidates is derived from EU directives and regulations, Brexit and the new Trade and Cooperation Agreement between the European Union and the U.K. that took provisional effect on January 1, 2021 could materially impact the regulatory regime with respect to the development, manufacture, importation, approval and commercialization of our product candidates in the U.K. or the EU. For example, the U.K. is no longer be covered by the centralized procedures for obtaining EU-wide marketing authorization from the European Medicines Agency and a separate process for authorization of drug products, including our product candidates, will be required in the U.K. It is currently unclear whether the Medicines & Healthcare products Regulatory Agency in the U.K. is sufficiently prepared to handle the increased volume of marketing authorization applications that it is likely to receive. Any delay in obtaining, or an inability to obtain, any marketing approvals, as a result of Brexit or otherwise, would prevent us from commercializing our product candidates in the U.K. or the EU and restrict our ability to generate revenue and achieve and sustain profitability. In addition, we may be required to pay taxes or duties or be subjected to other hurdles in connection with the importation of our product candidates into the EU, or we may incur expenses in establishing a manufacturing facility in the EU in order to circumvent such hurdles. If any of these outcomes occur, we may be forced to restrict or delay efforts to seek regulatory approval in the U.K. or the EU for our product candidates, or incur significant additional expenses to operate our business, which could significantly and materially harm or delay our ability to generate revenues or achieve profitability of our business. Any further changes in international trade, tariff and import/export regulations as a result of Brexit or otherwise may impose unexpected duty costs or other non-tariff barriers on us. These developments, or the perception that any of them could occur, may significantly reduce global trade and, in particular, trade between the affected nations and the U.K. It is also possible that Brexit may negatively affect our ability to attract and retain employees, particularly those from the EU.
We face potential product liability, and, if successful claims are brought against us, we may incur substantial liability and costs. If the use or misuse of our products or product candidates harm patients or is perceived to harm patients even when such harm is unrelated to our products or product candidates, our regulatory approvals could be revoked, suspended or otherwise negatively affected, and we could be subject to costly and damaging product liability claims.
We face the risk of incurring liabilities to clinical trial patients if they are injured as a result of their participation in our clinical trials. In the event we commercialize Renevia in the EU or in other countries that recognize the CE Mark, we will also face product liability risks associated with the use of Renevia by consumers. If any claims are made and if liability can be established, the amount of any liability we or our affiliates may incur, could exceed any insurance coverage in effect, and the amount of the liability could be material to our financial condition.
The use or misuse of our product candidates in clinical trials and the sale of any products for which we obtain marketing approval, including Renevia, exposes us to the risk of product liability claims. Product liability claims might be brought against us by consumers, healthcare providers, pharmaceutical companies or others selling or otherwise coming into contact with our products. There is a risk that our product candidates may induce adverse events. If we cannot successfully defend against product liability claims, we could incur substantial liability and costs. In addition, regardless of merit or eventual outcome, product liability claims may result in:
| ● | impairment of our business reputation; |
| ● | initiation of investigations by regulators; | |
| ● | withdrawal of clinical trial participants; | |
| ● | costs due to related litigation; | |
| ● | distraction of management’s attention from our primary business; | |
| ● | substantial monetary awards to patients or other claimants; | |
| ● | the inability to commercialize our product candidates; | |
| ● | product recalls, withdrawals or labeling, marketing or promotional restrictions; and | |
| ● | decreased demand for our product candidates, if approved for commercial sale. |
| 46 |
We believe our current product liability insurance coverage is appropriate in light of our clinical programs; however, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. If and when we obtain marketing approval for product candidates, we intend to increase our insurance coverage to include the sale of commercial products; however, we may be unable to obtain product liability insurance on commercially reasonable terms or in adequate amounts. Significant damages have been awarded in class action lawsuits based on drugs or medical treatments that had unanticipated adverse effects. A successful product liability claim or series of claims brought against us could cause our stock price to decline and, if the amount of damages exceeds our insurance coverage, could adversely affect our results of operations and business.
Cell Cure has received Israeli government grants for certain of its research and development activities. The terms of these grants may require Cell Cure to seek approvals and to satisfy specified conditions to manufacture products and transfer or license grant-supported technologies outside of Israel. In the context of such approvals, Cell Cure will be required to pay penalties in addition to the repayment of the grants. Such grants are applied for on a yearly basis and may not be available or only partially granted in the future, which would increase our costs.
Cell Cure has received Israeli government grants for certain of its research and development activities. The terms of these grants require prior approval and the satisfaction of specified conditions to manufacture products and transfer or license technologies outside of Israel.
Under the Encouragement of Research, Development and Technological Innovation in the Industry Law 5744-1984 (formerly known as the Law for the Encouragement of Research and Development in Industry 5744-1984), and the regulations, guidelines, rules, procedures and benefit tracks thereunder (collectively, the “Innovation Law”), annual research and development programs that meet specified criteria and are approved by a committee of the Israel Innovation Authority (“IIA”) are eligible for grants. The grants awarded are typically up to 50% of the project’s expenditures, as determined by the IIA committee and subject to the benefit track under which the grant was awarded. A company that receives a grant from the IIA (a “Grant Recipient”), is typically required to pay royalties to the IIA on income generated from products incorporating know-how developed using such grants (including income derived from services associated with such products) or on all revenues of the Grant Recipient (depending upon the terms of the approval letters issued by the IIA), until 100% of the U.S. dollar-linked grant plus annual LIBOR interest is repaid. In general, the rate of such royalties varies between 3% to 5%.
The obligation to pay royalties is contingent on actual revenues being generated from such products and services or actual revenues being generated by the Grant Recipient in general (as the case may be). In the absence of such revenues, no payment of royalties is required. It should be noted that the restrictions under the Innovation Law will continue to apply even after the repayment of such royalties in full by the Grant Recipient including restrictions on the sale, transfer or licensing to a foreign entity of know-how developed as part of the programs under which the grants were given.
The terms of the grants under the Innovation Law also (generally) require that the products developed as part of the programs under which the grants were given be manufactured in Israel and that the know-how developed thereunder may not be transferred outside of Israel, unless prior written approval is received from the IIA (such approval is not required for the transfer of a portion of the manufacturing capacity which does not exceed, in the aggregate, 10% of the portion declared to be manufactured outside of Israel in the applications for funding (in which case only notification is required), and additional payments are required to be made to IIA). It should be noted that this does not restrict the export of products that incorporate the funded know-how.
The Innovation Law restricts the ability to transfer or license know-how funded by IIA outside of Israel. Transfer of IIA-funded know-how outside of Israel requires prior approval and is subject to approval and payment of a redemption fee to the IIA calculated according to the relevant formulas provided under the Innovation Law. A transfer or license for the purpose of the Innovation Law are generally interpreted very broadly and include, inter alia, any actual sale or assignment of the IIA-funded know-how, any license to further develop or otherwise exploit the IIA-funded know-how or the products resulting from such IIA-funded know-how or any other transaction, which, in essence, constitutes a transfer of the IIA-funded know-how. Generally, a mere license solely to market or distribute products resulting from the IIA-funded know-how would not be deemed a transfer or license for the purpose of the Innovation Law.
Part of Cell Cure’s research and development efforts have been financed, partially, through grants that it has received from the IIA and when we acquired our holdings in Cell Cure, we undertook in writing, vis-à-vis the IIA, to abide by, and to ensure the abidance of Cell Cure to, the Innovation Law. We therefore must comply with the requirements of the Innovation Law and related regulations. As of December 31, 2020, we received approximately $15.4 million of such grants.
| 47 |
The restrictions under the Innovation Law may impair our ability to enter into agreements which involve IIA-funded products or know-how without the approval of IIA. We cannot be certain that any approval of IIA will be obtained on terms that are acceptable to us, or at all. We may not receive the required approvals should we wish to transfer or license IIA-funded know-how, manufacturing and/or development outside of Israel in the future. Furthermore, in the event that we undertake a transaction involving the transfer to a non-Israeli entity of know-how developed with IIA-funding pursuant to a merger or similar transaction, the consideration available to our shareholders may be reduced by the amounts we are required to pay to the IIA. Any approval, if given, will generally be subject to additional financial obligations. Failure to comply with the requirements under the Innovation Law may subject Cell Cure to mandatory repayment of grants received by it (together with interest and penalties), as well as expose its directors and management to criminal proceedings. In addition, the IIA may from time to time conduct royalty audits. Further grants may not be approved or reduced in the future, which would increase our costs. IIA approval is not required for the marketing or distribution of products resulting from the IIA-funded research or development in the ordinary course of business.
Our international business exposes us to business, regulatory, political, operational, financial and economic risks associated with doing business outside of the United States.
Cell Cure is our 99% owned subsidiary located in Jerusalem, Israel. OpRegen is currently manufactured at Cell Cure and we anticipate transitioning some or all of the manufacturing of OPC1 and VAC2 to Cell Cure as well. A portion of our OpRegen Phase 1/2a clinical trial has been conducted at sites in Israel. Conducting operations internationally involves a number of risks, including:
| ● | difficulty in staffing and managing foreign operations; | |
| ● | failure by us to obtain the appropriate regulatory approvals; | |
| ● | logistics and regulations associated with shipping drug product or patient samples, including infrastructure conditions and transportation delays; | |
| ● | financial risks, such as longer payment cycles and exposure to foreign currency exchange rate fluctuations; | |
| ● | political and economic instability, including wars, terrorism, and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions; | |
| ● | multiple, conflicting and changing laws and regulations such as tax laws, export and import restrictions, employment laws, data and privacy laws, regulatory requirements and other governmental approvals, permits and licenses; and | |
| ● | regulatory and compliance risks that may fall within the purview of the U.S. Foreign Corrupt Practices Act, UK Bribery Act, anti-boycott laws and other anti-corruption laws. |
Any of these factors could significantly harm our international operations and, consequently, our results of operations. In addition, any failure to comply with applicable legal and regulatory obligations could impact us in a variety of ways that include, but are not limited to, significant criminal, civil and administrative penalties, including imprisonment of individuals, fines and penalties, denial of export privileges, seizure of shipments, and restrictions on certain business activities. Also, the failure to comply with applicable legal and regulatory obligations could result in the disruption of our clinical trial activities.
Our international operations could be affected by changes in laws, trade regulations, labor and employment regulations, and procedures and actions affecting approval, production, pricing, reimbursement and marketing of tests, as well as by inter-governmental disputes. Any of these changes could adversely affect our business.
Our success internationally will depend, in part, on our ability to develop and implement policies and strategies that are effective in anticipating and managing these and other risks in Israel. Failure to manage these and other risks may have a material adverse effect on our operations in Israel and on our business as a whole.
| 48 |
Risks Related to our Intellectual Property
Our intellectual property may be insufficient to protect our products.
Our patents and patent applications are directed to compositions of matter, formulations, methods of use and/or methods of manufacturing, as appropriate. In addition to patenting our own technology and that of our subsidiaries, we have licensed patents and patent applications for certain stem cell technology, hEPC, and hES cell lines, hydrogel technology and other technology from other companies.
The patent positions of pharmaceutical and biotechnology companies, including ours, are generally uncertain and involve complex legal and factual questions. Our business could be negatively affected by any of the following:
| ● | the claims of any patents that are issued may not provide meaningful protection, may not provide a basis for commercially viable products or may not provide us with any competitive advantages; | |
| ● | our patents may be challenged by third parties; | |
| ● | others may have patents that relate to our technology or business that may prevent us from marketing our product candidates unless we are able to obtain a license to those patents; | |
| ● | the pending patent applications to which we have rights may not result in issued patents; | |
| ● | our patents may have terms that are inadequate to protect our competitive position on our products; | |
| ● | we may not be successful in developing additional proprietary technologies that are patentable. |
In addition, others may independently develop similar or alternative technologies, duplicate any of our technologies and, if patents are licensed or issued to us, design around the patented technologies licensed to or developed by us. As an example, Astellas’ patent portfolio with respect to the manufacture of its RPE products could adversely impact our rights to manufacture OpRegen. Moreover, we could incur substantial costs in litigation if we have to defend ourselves in patent lawsuits brought by third parties or if we initiate such lawsuits.
If we are unable to obtain and enforce patents and to protect our trade secrets, others could use our technology to compete with us, which could limit opportunities for us to generate revenues by licensing our technology and selling products.
Our success will depend in part on our ability to obtain and enforce patents and maintain trade secrets in the United States and in other countries. If we are unsuccessful at obtaining and enforcing patents, our competitors could use our technology and create products that compete with our products, without paying license fees or royalties to us. The preparation, filing, and prosecution of patent applications can be costly and time consuming. Our limited financial resources may not permit us to pursue patent protection of all of our technology and products in all key markets. Even if we are able to obtain issued patents covering our technology or products, we may have to incur substantial legal fees and other expenses to enforce our patent rights to protect our technology and products from infringing uses. We may not have the financial resources to finance the litigation required to preserve our patent and trade secret rights. Litigation, interferences, oppositions, inter partes reviews or other proceedings are, have been and may in the future be necessary in some instances to determine the validity and scope of certain of our proprietary rights, and in other instances to determine the validity, scope or non-infringement of certain patent rights claimed by third parties to be pertinent to the manufacture, use or sale of our products. This means that patents owned or licensed by us may be lost if the outcome of a proceeding is unfavorable to us.
There is no certainty that our pending or future patent applications will result in the issuance of patents.
Our success depends in part on our ability to obtain and defend patent and other intellectual property rights that are important to the commercialization of our products and product candidates. The degree of patent protection that will be afforded to our products and processes in the U.S. and in other important markets remains uncertain and is dependent upon the scope of protection decided upon by the patent offices, courts, administrative bodies and lawmakers in these countries. We can provide no assurance that we will successfully obtain or preserve patent protection for the technologies incorporated into our products and processes, or that the protection obtained will be of sufficient breadth and degree to protect our commercial interests in all countries where we conduct business. If we cannot prevent others from exploiting our inventions, we will not derive the benefit from them that we currently expect. Furthermore, we can provide no assurance that our products will not infringe patents or other intellectual property rights held by third parties.
| 49 |
In Europe, there is uncertainty about the eligibility of hES cell subject matter for patent protection. The European Patent Convention prohibits the granting of European patents for inventions that concern “uses of human embryos for industrial or commercial purposes.” A recent decision at the Court of Justice of the European Union interpreted parthenogenetically produced hES cells as patentable subject matter. Consequently, the European Patent Office now recognizes that human pluripotent stem cells (including human ES cells) can be created without a destructive use of human embryos as of June 5, 2003, and patent applications relating to hES cell subject matter with a filing and priority date after this date are no longer automatically excluded from patentability under Article 53 (a) EPC and Rule 28(c) EPC.
Intellectual property we may develop using grants received from governments are subject to rights maintained by those governments.
Research and development we perform that is funded by grants from government, and any intellectual property that we create using those grants, is subject to certain rights of the government entities to require that we license or grant rights to the intellectual property developed using government funding in certain circumstances.
There is no certainty that we will be able to obtain licenses to intellectual property rights owned by third parties.
There are no assurances that any of our intellectual property rights will guarantee protection or market exclusivity for our products and product candidates. In such cases, we may need to obtain enabling licenses from third parties to protect our products and product candidates, try to secure market exclusivity or avoid infringing on the intellectual property rights of third parties. If we are unable to fully protect our product candidates or achieve market exclusivity for our products and product candidates, our financial success will be dependent, in part, on our ability to protect and enforce our intellectual property rights, to operate without infringing upon the proprietary rights of others, or, when necessary, our ability to obtain enabling licenses.
If we fail to meet our obligations under license agreements, we may lose our rights to key technologies on which our business depends.
Our business depends on several critical technologies that are based in part on technology licensed from third parties. Those third-party license agreements impose obligations on us, including payment obligations and obligations to pursue development of commercial products under the licensed patents or technology. If a licensor believes that we have failed to meet our obligations under a license agreement, the licensor could seek to limit or terminate our license rights, which could lead to costly and time-consuming litigation and, potentially, a loss of the licensed rights. During the period of any such litigation, our ability to carry out the development and commercialization of potential products, and our ability to raise any capital that we might then need, could be significantly and negatively affected. If our license rights were restricted or ultimately lost, we would not be able to continue to use the licensed technology in our business.
Risks Related to our Dependence on Third Parties
We may become dependent on possible future collaborations to develop and commercialize many of our product candidates and to provide the regulatory compliance, sales, marketing and distribution capabilities required for the success of our business.
We may enter into various kinds of collaborative research and development and product marketing agreements to develop and commercialize our products. The expected future milestone payments and cost reimbursements from collaboration agreements could provide an important source of financing for our research and development programs, thereby facilitating the application of our technology to the development and commercialization of our products, but there are risks associated with entering into collaboration arrangements.
| 50 |
There is a risk we could become dependent upon one or more collaborative arrangements. A collaborative arrangement upon which we might depend might be terminated by our collaboration partner or a partner might determine not to actively pursue the development or commercialization of our products. A collaboration partner also may not be precluded from independently pursuing competing products and drug delivery approaches or technologies.
There is a risk that a collaboration partner might fail to perform its obligations under the collaborative arrangements or may be slow in performing its obligations. In addition, a collaboration partner may experience financial difficulties at any time that could prevent it from having available funds to contribute to the collaboration. If a collaboration partner fails to conduct its product development, commercialization, regulatory compliance, sales and marketing or distribution activities successfully and in a timely manner, or if it terminates or materially modifies its agreements with us, the development and commercialization of one or more product candidates could be delayed, curtailed or terminated because we may not have sufficient financial resources or capabilities to continue such development and commercialization on our own.
We do not have the ability to independently conduct clinical trials required to obtain regulatory approvals for our product candidates.
We will need to rely on third parties, such as CROs, data management companies, contract clinical research associates, medical institutions, clinical investigators and contract laboratories to conduct any clinical trials we may undertake for our product candidates. We may also rely on third parties to assist with preclinical development of our product candidates. If we outsource clinical trials, we may not directly control the timing, conduct and expense of our clinical trials. If we enlist third parties to conduct clinical trials and they fail to perform their contractual duties or regulatory obligations or fail to meet expected deadlines, if they need to be replaced or if the quality or accuracy of the data they obtain is compromised due to failing to adhere to our clinical protocols or regulatory requirements or for other reasons, our preclinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not obtain regulatory approval for or successfully commercialize our product candidates.
In addition, quarantines, shelter-in-place and similar government orders, or the perception that such orders, shutdowns or other restrictions on the conduct of business operations could occur, related to COVID-19 or other infectious diseases could impact personnel at these third parties, which could disrupt our clinical timelines, which could have a material adverse impact on our business, prospects, financial condition and results of operations.
We have relied on CIRM to fund past clinical trials of OPC1 and we do not know if they will provide additional funding for future studies of OPC1.
We received $14.3 million of funding from CIRM to support clinical development of OPC1. We intend to apply for additional CIRM grants, if available; however, we cannot provide any assurance that such grants will be awarded. If we are unable to obtain another CIRM grant, we will need to raise funds through other mechanisms to support future clinical studies of OPC1, which may take additional time and effort. If capital is not immediately available, this may force us to amend, delay, or discontinue the clinical trial and development work for OPC1 until funding is secured.
We may need to rely on marketing partners or contract sales companies.
If we are able to develop our product candidates and obtain necessary regulatory approvals, we may need to rely on marketing, selling or distributing partners. If we do not partner for commercial services, we will depend on our ability to build our own marketing, selling and distribution capabilities, which would require the investment of significant financial and management resources, or we will need to find collaborative marketing partners, sales representatives or wholesale distributors for the commercial sale of our products.
If we market products through arrangements with third parties, we may pay sales commissions to sales representatives or we may sell or consign products to distributors at wholesale prices. As a result, our gross profit from product sales may be lower than it would be if we sold our products directly to end users at retail prices through our own sales force. There can be no assurance we will be able to negotiate distribution or sales agreements with third parties on favorable terms to justify our investment in our products or achieve sufficient revenues to support our operations.
| 51 |
Risks Pertaining to Our Common Shares
Because we are engaged in the development of pharmaceutical and stem cell therapy products, the price of our common shares may rise and fall rapidly.
The market price of our common shares, like that of the shares of many biotechnology companies, has been highly volatile. The price of our common shares may rise rapidly in response to certain events, such as the commencement of clinical trials of an experimental new therapy, even though the outcome of those trials and the likelihood of ultimate FDA approval of a therapeutic product remain uncertain. Similarly, prices of our common shares may fall rapidly in response to certain events such as unfavorable results of clinical trials or a delay or failure to obtain FDA approval. The failure of our earnings to meet analysts’ expectations could result in a significant rapid decline in the market price of our common shares.
Current economic and stock market conditions may adversely affect the price of our common shares.
The stock market has been experiencing extreme price and volume fluctuations which have affected the market price of the equity securities without regard to the operating performance of the issuing companies. Broad market fluctuations, as well as general economic, political and other conditions (such as the recent coronavirus outbreak), may adversely affect the market price of our common shares.
Because we do not pay cash dividends, our common shares may not be a suitable investment for anyone who needs to earn dividend income.
We do not pay cash dividends on our common shares. For the foreseeable future, we anticipate that any earnings generated in our business will be used to finance the growth of our business and will not be paid out as dividends to holders of our common shares. This means that our common shares may not be a suitable investment for anyone who needs to earn income from their investments.
Insiders continue to have substantial influence over our company, which could limit your ability to influence the outcome of key transactions, including a change of control.
Our directors, executive officers and their affiliates, in the aggregate, owned approximately 27% of our outstanding common shares as of December 31, 2020. As a result, these shareholders, if acting together, will be able to heavily influence or control matters requiring approval by our shareholders, including the election of directors and the approval of mergers, acquisitions or other extraordinary transactions. They may also have interests that differ from yours and may vote in a way with which you disagree, and which may be averse to your interests. This concentration of ownership may have the effect of delaying, preventing or deterring a change of control of our company, could deter certain public investors from purchasing our common shares and might ultimately affect the market price of our common shares.
Our business could be negatively affected as a result of actions of activist shareholders, and such activism could affect the trading value of our securities.
Shareholders may, from time to time, engage in proxy solicitations or advance stockholder proposals, or otherwise attempt to effect changes and assert influence on our board of directors and management. Activist campaigns that contest or conflict with our strategic direction or seek changes in the composition of our board of directors could have an adverse effect on our operating results and financial condition. A proxy contest would require us to incur significant legal and advisory fees, proxy solicitation expenses and administrative and associated costs and require significant time and attention by our board of directors and management, diverting their attention from the pursuit of our business strategy. Any perceived uncertainties as to our future direction and control, our ability to execute on our strategy, or changes to the composition of our board of directors or senior management team arising from a proxy contest could lead to the perception of a change in the direction of our business or instability which may result in the loss of potential business opportunities, make it more difficult to pursue our strategic initiatives, or limit our ability to attract and retain qualified personnel and business partners, any of which could adversely affect our business and operating results. If individuals are ultimately elected to our board of directors with a specific agenda, it may adversely affect our ability to effectively implement our business strategy and create additional value for our stockholders. We may choose to initiate, or may become subject to, litigation as a result of the proxy contest or matters arising from the proxy contest, which would serve as a further distraction to our board of directors and management and would require us to incur significant additional costs. In addition, actions such as those described above could cause significant fluctuations in our stock price based upon temporary or speculative market perceptions or other factors that do not necessarily reflect the underlying fundamentals and prospects of our business.
| 52 |
Securities analysts may not initiate coverage or continue to cover our common shares, and this may have a negative impact on the market price of our common shares.
The trading market for our common shares depends, in part, on the research and reports that securities analysts publish about our business and our common shares. We do not have any control over these analysts. There is no guarantee that securities analysts will cover our common shares. If securities analysts do not cover our common shares, the lack of research coverage may adversely affect the market price of those shares. If securities analysts do cover our common shares, they could issue reports or recommendations that are unfavorable to the price of our common shares, and they could downgrade a previously favorable report or recommendation, and in either case our share prices could decline as a result of the report. If one or more of these analysts does not initiate coverage, ceases to cover our common shares or fails to publish regular reports on our business, we could lose visibility in the financial markets, which could cause our share prices or trading volume to decline.
If we or our subsidiaries issue additional common shares or preferred shares, investors in our common shares may experience dilution of their ownership interests.
We and our subsidiaries may issue additional common shares or other securities convertible into or exercisable for common shares to raise additional capital or to hire or retain employees or consultants, or in connection with future acquisitions of companies or licenses to technology or rights, or for other business purposes. The future issuance of additional securities may be dilutive to our shareholders and may create downward pressure on the trading price of our common shares.
We are currently authorized to issue an aggregate of 252,000,000 shares of capital stock consisting of 250,000,000 common shares and 2,000,000 “blank check” preferred shares, which means we may issue, without stockholder approval, one or more series of preferred stock having such designation, powers, privileges, preferences, including preferences over our common shares respecting dividends and distributions, terms of redemption and relative participation, optional, or other rights, if any, of the shares of each such series of preferred stock and any qualifications, limitations or restrictions thereof, as our board of directors may determine. The terms of one or more series of preferred stock could dilute the voting power or reduce the value of our common shares. Any preferred shares may also be convertible into common shares on terms that would be dilutive to holders of common shares. Our subsidiaries may also issue their own preferred shares with a similar impact on our ownership of the subsidiaries.
As of December 31, 2020, Lineage had 153,095,883 common shares outstanding, 16,214,547 common shares reserved for issuance upon the exercise of outstanding options under our employee stock option plans, 92,700 common shares reserved for issuance upon the vesting and settlement of restricted stock units under our equity incentive plan, and 1,089,900 common shares subject to warrants.
In addition, in May 2020 we entered into a Controlled Equity OfferingSM Sales Agreement (the “Sales Agreement”) with Cantor Fitzgerald & Co., as sales agent (“Cantor Fitzgerald”), pursuant to which we may, but are not obligated to, raise up to $25.0 million through the sale of common shares from time to time in at-the-market transactions under the Sales Agreement. As of December 31, 2020, we made $5.1 million in sales under the Sales Agreement (which excludes $0.3 million of cash in transit related to 2020 sales that settled in 2021).
The operation of some of our subsidiaries has been financed in part through the sale of shares of capital stock and warrants to purchase securities of those subsidiaries to private investors. Future sales of such securities by our subsidiaries could reduce our ownership interest in the applicable subsidiary, and correspondingly dilute our shareholder’s ownership interests in our consolidated enterprise. Certain of our subsidiaries also have their own stock option plans and the exercise of stock options or the sale of restricted stock under those plans would also reduce our ownership interest in the applicable subsidiary, with a resulting dilutive effect on the ownership interest of our shareholders in our consolidated enterprise.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
| 53 |
| ITEM 2. | PROPERTIES |
General
In general, we believe that our properties are well-maintained, adequate and suitable for their current requirements and for our operations in the foreseeable future. See the Notes to Consolidated Financial Statements – Note 14. Commitments and Contingencies included elsewhere in this Report.
Lineage Facilities
Our corporate headquarters comprise 8,841 square feet of rentable space in an office park in Carlsbad, California. We also sublease 2,432 square feet of space in Alameda, California.
Cell Cure Facilities
Cell Cure leases 728.5 square meters (approximately 7,842 square feet) of office and laboratory space in the Bio Park on the campus of the Hadassah University Hospital in Jerusalem, Israel under a lease that expires on December 31, 2025. We have an option to extend the term for an additional 5 years.
In January 2018, Cell Cure entered into another lease for an additional 934 square meters (approximately 10,054 square feet) of office space in the same facility in Jerusalem, Israel under a lease that expires on December 31, 2025, with two options to extend the lease for 5 years each. The term of this lease commenced on April 1, 2018 and includes a leasehold improvement construction allowance of up to NIS 4,000,000 (approximately up to $1.1 million) from the landlord. The leasehold improvements were substantially completed by December 31, 2018 and the construction allowance was fully utilized.
| ITEM 3. | LEGAL PROCEEDINGS |
From time to time, we are subject to legal proceedings and claims in the ordinary course of business. While management presently believes that the ultimate outcome of these proceedings, individually and in the aggregate, will not materially harm our financial position, cash flows, or overall trends in results of operations, legal proceedings are subject to inherent uncertainties, and unfavorable rulings or outcomes could occur that have individually or in aggregate, a material adverse effect on our business, financial condition or operating results. Except as described below, we are not currently subject to any pending material litigation, other than ordinary routine litigation incidental to our business, as described above.
On February 19, 2019, a putative shareholder class action lawsuit was filed (captioned Lampe v. Asterias Biotherapeutics, Inc. et al., Case No. RG19007391) in the Superior Court of the State of California, County of Alameda challenging the Asterias Merger. On March 1, 2019, Asterias made certain amendments and supplements to its public disclosures regarding the Asterias Merger (the “Supplemental Disclosures”). On May 3, 2019, an amended class action complaint (the “Amended Complaint”) was filed. The Amended Complaint named Lineage, Patrick Merger Sub, Inc., the Asterias board of directors, one member of Lineage’s board of directors, and certain stockholders of both Lineage and Asterias. The action was brought by two purported stockholders of Asterias, on behalf of a putative class of Asterias stockholders, and asserted breach of fiduciary duty and aiding and abetting claims under Delaware law. The Amended Complaint alleged, among other things, that the process leading up to the Asterias Merger was conflicted and inadequate, and that the proxy statement filed by Asterias with the Commission omitted certain material information, which allegedly rendered the information disclosed materially misleading. The Amended Complaint sought, among other things, that a class be certified, the recovery of monetary damages, and attorneys’ fees and costs.
On June 3, 2019, defendants filed demurrers to the Amended Complaint. On August 13, 2019, the parties submitted a stipulation to the court seeking dismissal of the action with prejudice as to the named Plaintiffs and without prejudice as to the unnamed putative class members, and disclosing to the court the parties’ agreement to resolve, for $200,000, Plaintiffs’ claim for an award of attorneys’ fees and expenses in connection with the purported benefit conferred on Asterias stockholders by the Supplemental Disclosures. The court granted the stipulation and dismissed the action August 14, 2019. Lineage continues to believe that the claims and allegations in the action lack merit, but believed that it was in Lineage’s shareholders’ best interest for the action to be dismissed and to resolve the fee claim in a timely manner without additional costly litigation expenses.
On October 14, 2019, another putative class action lawsuit was filed challenging the Asterias Merger. This action (captioned Ross v. Lineage Cell Therapeutics, Inc., et al., C.A. No. 2019-0822) was filed in Delaware Chancery Court and names Lineage, the Asterias board of directors, one member of Lineage’s board of directors, and certain stockholders of both Lineage and Asterias as defendants. The action was brought by a purported stockholder of Asterias, on behalf of a putative class of Asterias stockholders, and asserts breach of fiduciary duty and aiding and abetting claims under Delaware law. The complaint alleges, among other things, that the process leading up to the Asterias Merger was conflicted, that the Asterias Merger consideration was inadequate, and that the proxy statement filed by Asterias with the Commission omitted certain material information, which allegedly rendered the information disclosed materially misleading. The complaint seeks, among other things, that a class be certified, the recovery of monetary damages, and attorneys’ fees and costs. On December 20, 2019, the defendants moved to dismiss the complaint. On February 10, 2020, the plaintiff filed an opposition. Defendants filed their replies on March 13, 2020. On June 23, 2020, a hearing on the motions to dismiss occurred. On September 21, 2020, the Chancery Court denied the motion to dismiss as to Lineage and certain members of the Asterias board of directors, and it granted the motion to dismiss as to all other defendants. On October 30, 2020, the remaining defendants filed an answer to the complaint.
Lineage believes the allegations in the action lack merit and intends to vigorously defend the claims asserted. It is impossible at this time to assess whether the outcome of this proceeding will have a material adverse effect on Lineage’s consolidated results of operations, cash flows or financial position. Therefore, in accordance with ASC 450, Contingencies, Lineage has not recorded any accrual for a contingent liability associated with this legal proceeding based on its belief that a liability, while possible, is not probable nor estimable, and any range of potential contingent liability amounts cannot be reasonably estimated at this time. Lineage records legal expenses as incurred.
| ITEM 4. | MINE SAFETY DISCLOSURES |
Not applicable.
| 54 |
PART II
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS, AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our common shares trade on the NYSE American and on the Tel Aviv Stock Exchange under the ticker symbol LCTX.
Holders
As of March 1, 2021, there were 384 record holders of our common shares. The number of beneficial owners is substantially greater than the number of record holders because a large portion of our common shares is held of record through brokerage firms in “street name.”
Dividend Policy
We have not paid dividends on our common shares. We currently intend to retain any earnings for use in the operations of our business. We, therefore, do not anticipate paying cash dividends on our common shares in the foreseeable future.
Recent Sales of Unregistered Securities
Except as previously reported in our quarterly reports on Form 10-Q and current reports on Form 8-K filed with the Securities and Exchange Commission, during the year ended December 31, 2020, there were no unregistered sales of equity securities by us during the year ended December 31, 2020.
| ITEM 6. | SELECTED FINANCIAL DATA |
We are a smaller reporting company as defined by Rule 12b-2 of the Securities Exchange Act of 1934, as amended. Accordingly, we are not required to provide the information required by this item in this Report.
| 55 |
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations is intended to provide information necessary to understand our audited consolidated financial statements for the two-year period ended December 31, 2020, and highlight certain other information which, in the opinion of management, will enhance a reader’s understanding of our financial condition, changes in financial condition and results of operations. In particular, the discussion is intended to provide an analysis of significant trends and material changes in our financial position and the operating results of our business during the year ended December 31, 2020 as compared to the year ended December 31, 2019. This discussion should be read in conjunction with our consolidated financial statements and related notes included elsewhere in this Report. These historical financial statements may not be indicative of our future performance. This Management’s Discussion and Analysis of Financial Condition and Results of Operations contains a number of forward-looking statements, all of which are based on our current expectations and could be affected by the uncertainties and risks described throughout this Report, particularly in “Item 1A. Risk Factors.”
Overview
We are a clinical-stage biotechnology company developing novel cell therapies for unmet medical needs. Our focus is to develop therapies for degenerative retinal diseases, neurological conditions associated with demyelination, and aiding the body in detecting and combating cancer. Specifically, we are testing therapies to treat dry age-related macular degeneration (“AMD”), spinal cord injuries, and non-small cell lung cancer. Our programs are based on our proprietary cell-based therapy platform and associated development and manufacturing capabilities. From this platform, we develop and manufacture specialized, terminally or functionally differentiated human cells from established and well-characterized pluripotent cell lines. These differentiated cells are developed either to replace or support cells that are dysfunctional or absent due to degenerative disease or traumatic injury, or are administered as a means of helping the body mount an effective immune response to cancer.
We have three allogeneic, or “off-the-shelf,” cell therapy programs in clinical development:
| ● | OpRegen®, a retinal pigment epithelium cell replacement therapy currently in a Phase 1/2a multicenter clinical trial for the treatment of advanced dry AMD with geographic atrophy. There currently are no therapies approved by the U.S. Food and Drug Administration (“FDA”) for dry AMD, which accounts for approximately 85-90% of all AMD cases and is the leading cause of blindness in people over the age of 60. | |
| ● | OPC1, an oligodendrocyte progenitor cell therapy currently in a Phase 1/2a multicenter clinical trial for acute spinal cord injuries. This clinical trial has been partially funded by the California Institute for Regenerative Medicine. | |
| ● | VAC2, an allogeneic cancer immunotherapy of antigen-presenting dendritic cells currently in a Phase 1 clinical trial in non-small cell lung cancer. This clinical trial is being funded and conducted by Cancer Research UK, the world’s largest independent cancer research charity. |
Lineage completed its merger (the “Asterias Merger”) with Asterias Biotherapeutics, Inc. (“Asterias”) on March 8, 2019, which incorporated OPC1 and VAC2 into its cell therapy product portfolio.
In addition to seeking to create value for shareholders by developing product candidates and other technologies through our clinical development programs, we also seek to create value from our technologies through partnering and strategic transactions. We founded two companies that later became publicly traded companies: OncoCyte Corporation (“OncoCyte”) and AgeX Therapeutics, Inc. (“AgeX”).
During the year ended December 31, 2020, we received approximately $12.6 million in gross proceeds in connection with our sale of shares of OncoCyte and AgeX. In August 2020, we also received $24.6 million from Juvenescence Limited (“Juvenescence”), representing principal and accrued interest under a promissory note we received in connection with our sale of AgeX shares to Juvenescence in August 2018.
We no longer hold any common stock in AgeX. The value of our OncoCyte holdings as of March 5, 2021, was approximately $4.2 million, based on the closing price of its common stock on that date. In this Report, see Part I, Item 1A, “Risk Factors—Risks Related to Our Business Operations and Capital Requirements—The value of our investments in public companies fluctuates based on their respective stock prices and could be negatively affected by poor business performance.”
| 56 |
Though our principal focus is on advancing our three cell therapy programs in clinical development, we may seek to create additional value through corporate transactions, as we have in the past.
Critical Accounting Policies
The preparation of consolidated financial statements in conformity with accounting principles generally accepted in the United States (“GAAP”) requires management to make estimates and assumptions that affect the reported amounts in our consolidated financial statements and related notes. Our significant accounting policies are described in Note 2 to our consolidated financial statements included elsewhere in this Report. We have identified below our critical accounting policies and estimates that we believe require the greatest amount of judgment. On an ongoing basis, we evaluate estimates which are subject to significant judgment, including those related to going concern assessment of our consolidated financial statements, useful lives associated with long-lived assets, including evaluation of asset impairment, allowances for uncollectible accounts and financing receivables, valuing shares owned in nonconsolidated companies using the equity method of accounting, loss contingencies, deferred income taxes and tax reserves, including valuation allowances related to deferred income taxes, and assumptions used to value stock-based awards, debt or other equity instruments. Actual results could differ materially from those estimates. On an ongoing basis, we evaluate our estimates compared to historical experience and trends which form the basis for making judgments about the carrying value of assets and liabilities. To the extent that there are material differences between our estimates and our actual results, our future financial statement presentation, financial condition, results of operations and cash flows will be affected.
We believe the assumptions and estimates associated with the following have the greatest potential impact on our consolidated financial statements.
Business Combinations – We account for business combinations, such as the Asterias Merger, in accordance with Accounting Standards Codification (“ASC”) Topic 805, Business Combinations, which requires the purchase price to be measured at fair value. When the purchase consideration consists entirely of our common shares, we calculate the purchase price by determining the fair value, as of the acquisition date, of shares issued in connection with the closing of the acquisition. We recognize estimated fair values of the tangible assets and intangible assets acquired, including in-process research and development (“IPR&D”), and liabilities assumed as of the acquisition date, and we record as goodwill any amount of the fair value of the tangible and intangible assets acquired and liabilities assumed in excess of the purchase price.
Goodwill and IPR&D – Goodwill is calculated as the difference between the acquisition date fair value of the consideration transferred and the values assigned to the assets acquired and liabilities assumed. Goodwill is not amortized but is tested for impairment at least annually, or more frequently if circumstances indicate potential impairment. IPR&D assets are indefinite-lived intangible assets until the completion or abandonment of the associated research and development (“R&D”) efforts. Once the R&D efforts are completed or abandoned, the IPR&D will either be amortized over the asset life as a finite-lived intangible asset or be impaired, respectively, in accordance with ASC 350, Intangibles – Goodwill and Other. In accordance with ASC 350, goodwill and acquired IPR&D are determined to have indefinite lives and, therefore, are not amortized. Instead, they are tested for impairment at least annually and between annual tests if we become aware of an event or a change in circumstances that would indicate the asset may be impaired.
Leases – We account for leases in accordance with ASC 842, Leases. We determine if an arrangement is a lease at inception. Leases are classified as either financing or operating, with classification affecting the pattern of expense recognition in the consolidated statements of operations. Under the available practical expedients for the adoption of ASC 842, we account for the lease and non-lease components as a single lease component. We recognize right-of-use (“ROU”) assets and lease liabilities for leases with terms greater than twelve months in the consolidated balance sheet. ROU assets represent our right to use an underlying asset during the lease term and lease liabilities represent our obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized at commencement date based on the present value of lease payments over the lease term. As most of our leases do not provide an implicit rate, we use our incremental borrowing rate based on the information available at commencement date in determining the present value of lease payments. We use the implicit rate when readily determinable. The operating lease ROU asset also includes any lease payments made and excludes lease incentives. Our lease terms may include options to extend or terminate the lease when it is reasonably certain that we will exercise that option. Lease expense for lease payments is recognized on a straight-line basis over the lease term. Operating leases are included as right-of-use assets in property and equipment, and ROU lease liabilities, current and long-term, in the consolidated balance sheets. Financing leases are included in property and equipment, and in financing lease liabilities, current and long-term, in the consolidated balance sheets. We disclose the amortization of our ROU assets and operating lease payments as a net amount, “Amortization of ROU assets”, on the consolidated statement of cash flows.
| 57 |
Going concern assessment – In accordance with Accounting Standards Update (“ASU”) 2014-15, Presentation of Financial Statements – Going Concern, we assess going concern uncertainty in our consolidated financial statements to determine if we have sufficient cash and cash equivalents on hand and working capital to operate for a period of at least one year from the date our consolidated financial statements are issued or are available to be issued, which is referred to as the “look-forward period” as defined by ASU No. 2014-15. As part of this assessment, based on conditions that are known and reasonably knowable to us, we will consider various scenarios, forecasts, projections, and estimates, and we will make certain key assumptions, including the timing and nature of projected cash expenditures or programs, and our ability to delay or curtail those expenditures or programs, if necessary, among other factors. Based on this assessment, as necessary or applicable, we make certain assumptions concerning our ability to curtail or delay research and development programs and expenditures to the extent we deem probable those implementations can be achieved and we have the proper authority to execute them within the look-forward period in accordance with ASU 2014-15.
Marketable Equity Securities – We account for our shares in OncoCyte and HBL (and previously AgeX) as marketable equity securities in accordance with ASC 320-10-25, Investments – Debt and Equity Securities, as amended by Accounting Standards Update (“ASU”) 2016-01, Financial Instruments–Overall: Recognition and Measurement of Financial Assets and Financial Liabilities, further discussed below.
OncoCyte and AgeX shares have readily determinable fair values quoted on the NYSE American under trading symbols “OCX” and “AGE”. The HBL shares have a readily determinable fair value quoted on the Tel Aviv Stock Exchange (“TASE”) under trading symbol “HDST” where share prices are denominated in New Israeli Shekels (NIS).
Prior to September 11, 2019, we accounted for our OncoCyte shares held at fair value, using the equity method of accounting. On September 11, 2019, Lineage’s ownership percentage decreased from 24% to 16% when it sold 4.0 million shares of OncoCyte common stock. Accordingly, as the ownership percentage was reduced to less than 20%, we are no longer considered to exercise significant influence over OncoCyte and are now accounting for our OncoCyte holdings as marketable equity securities. Prior to the Asterias Merger completed on March 8, 2019, we accounted for our Asterias shares held at fair value, using the equity method of accounting.
Royalties from product sales and license fees – Lineage’s performance obligations in agreements with certain customers is to provide a license to allow customers to make, import and sell company licensed products or methods for preclinical studies and commercial use. Customers pay a combination of a license issue fee paid up front and a sales-based royalty, if any, in some cases with yearly minimums. The transaction price is deemed to be the license issue fee stated in the contract. The license offered by Lineage is a functional license with significant standalone functionality and provides customers with the right to use Lineage’s intellectual property. This allows Lineage to recognize revenue on the license issue fee at a point in time at the beginning of the contract, which is when the customer begins to have use of the license. Variable consideration related to sales-based royalties is recognized only when (or as) the later of one or more of the following events occur: (i) a sale or usage occurs; or (ii) the performance obligation to which some, or all, of the sales-based or usage-based royalty that has been allocated and has been satisfied or partially satisfied. Due to the contract termination clauses, Lineage does not expect to receive all of the minimum royalty payments throughout the term of the agreements. Therefore, Lineage fully constrains recognition of the minimum royalty payments as revenues until its customers are obligated to pay, which is generally within 60 days prior to the beginning of each year the minimum royalty payments are due.
Grant revenues – In applying the provisions of Topic 606, Lineage has determined that government grants are out of the scope of Topic 606 because the government entities do not meet the definition of a “customer”, as defined by Topic 606, as there is not considered to be a transfer of control of good or services to the government entities funding the grant. Lineage has, and will continue to, account for grants received to perform research and development services in accordance with ASC 730-20, Research and Development Arrangements, which requires an assessment, at the inception of the grant, of whether the grant is a liability or a contract to perform research and development services for others. If Lineage or a subsidiary receiving the grant is obligated to repay the grant funds to the grantor regardless of the outcome of the research and development activities, then Lineage is required to estimate and recognize that liability. Alternatively, if Lineage or a subsidiary receiving the grant is not required to repay, or if it is required to repay the grant funds only if the research and development activities are successful, then the grant agreement is accounted for as a contract to perform research and development services for others, in which case, grant revenue is recognized when the related research and development expenses are incurred.
| 58 |
Deferred grant revenues represent grant funds received from the governmental funding agencies for which the allowable expenses have not yet been incurred as of the balance sheet date reported.
Long-lived intangible assets – Long-lived intangible assets, consisting primarily of acquired patents, patent applications, and licenses to use certain patents are stated at acquired cost, less accumulated amortization. Amortization expense is computed using the straight-line method over the estimated useful lives of the assets, generally over five to ten years.
Impairment of long-lived assets – Our long-lived assets, including long-lived intangible assets, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be fully recoverable. If an impairment indicator is present, we evaluate recoverability by a comparison of the carrying amount of the assets to future undiscounted net cash flows expected to be generated by the assets. If the assets are impaired, the impairment recognized is measured by the amount by which the carrying amount exceeds the estimated fair value of the assets.
Research and development – Research and development expenses consist of costs incurred for company-sponsored, collaborative and contracted research and development activities. These costs include direct and research-related overhead expenses including compensation and related benefits, stock-based compensation, consulting fees, research and laboratory fees, rent of research facilities, amortization of intangible assets, and license fees paid to third parties to acquire patents or licenses to use patents and other technology. We expense research and development costs as incurred. Research and development expenses incurred and reimbursed by grants from third parties approximate the grant income recognized in the consolidated statements of operations.
Stock-based compensation – We follow accounting standards governing share-based payments, which require the measurement and recognition of compensation expense for all share-based compensation awards made to directors and employees, including employee stock options, based on estimated fair values. We utilize the Black-Scholes option pricing model. Our determination of fair value of share-based payment awards on the date of grant using an option-pricing model is affected by our stock price as well as assumptions regarding a number of complex and subjective variables. These variables include, but are not limited to, expected stock price volatility over the term of the awards, and the expected term of options granted, which is derived using the simplified method, which is an average of the contractual term of the option and its vesting period, as we do not have sufficient historical exercise data. The risk-free rate is based on the U.S. Treasury yield in effect at the time of grant for zero coupon U.S. Treasury notes with maturities similar to the expected term of the awards. Forfeitures are accounted for as they occur.
Although the fair value of employee stock options is determined in accordance with FASB guidance, changes in the assumptions can materially affect the estimated value and therefore the amount of compensation expense recognized in the consolidated financial statements.
In management’s opinion, the existing valuation models may not provide an accurate measure of the fair value of employee stock options because the option-pricing model value may not be indicative of the fair value that would be established in a willing buyer/willing seller market transaction.
Income taxes – We account for income taxes in accordance with ASC 740, Income Taxes, which prescribe the use of the asset and liability method, whereby deferred tax asset or liability account balances are calculated at the balance sheet date using current tax laws and rates in effect. Valuation allowances are established when necessary to reduce deferred tax assets when it is more likely than not that a portion or all of the deferred tax assets will not be realized. ASC 740 guidance also prescribes a recognition threshold and a measurement attribute for the financial statement recognition and measurement of tax positions taken or expected to be taken in a tax return. For benefits to be recognized, a tax position must be more-likely-than-not sustainable upon examination by taxing authorities. We file a U.S. federal income tax return as well as various state and foreign income tax returns. Our judgments regarding future taxable income may change over time due to changes in market conditions, changes in tax laws, tax planning strategies or other factors. If our assumptions, and consequently the estimates, change in the future with respect to our own deferred tax assets and liabilities, the valuation allowance may be increased or decreased, which may have a material impact on our consolidated financial statements. We recognize accrued interest and penalties related to unrecognized tax benefits, if any, as income tax expense, however, no amounts were accrued for the payment of interest and penalties as of December 31, 2020 and 2019.
| 59 |
Principles of consolidation – Our consolidated financial statements include the accounts of our wholly owned and majority-owned subsidiaries. All material intercompany accounts and transactions have been eliminated in consolidation. The consolidated financial statements are presented in accordance with accounting principles generally accepted in the U.S. and with the accounting and reporting requirements of SEC Regulation S-X.
Results of Operations
Comparison of Years Ended December 31, 2020 and 2019
Revenues
The following table shows our revenues for the years ended December 31, 2020 and 2019 (amounts in thousands except percentages).
| Year Ended December 31, | $ Increase/ | % Increase/ | ||||||||||||||
| 2020 | 2019 | (Decrease) | (Decrease) | |||||||||||||
| Grant revenues | $ | 1,053 | $ | 2,037 | $ | (984 | ) | (48 | %) | |||||||
| Royalties from product sales and license fees | 773 | 1,221 | (448 | ) | (37 | %) | ||||||||||
| Sale of research products and services | - | 257 | (257 | ) | (100 | %) | ||||||||||
| Total revenues | 1,826 | 3,515 | (1,689 | ) | (48 | %) | ||||||||||
| Cost of sales | (385 | ) | (412 | ) | (27 | ) | (7 | %) | ||||||||
| Gross profit | $ | 1,441 | $ | 3,103 | $ | (1,662 | ) | (54 | %) | |||||||
Total revenues for the year ended December 31, 2020 were $1.8 million compared to $3.5 million for the year ended December 31, 2019. The decrease of $1.7 million is primarily due to a $1.0 million decrease in grant revenue, a $0.4 million decrease in royalties from product sales and license fees and a $0.3 million decrease in the sale of research products and services due to the cessation of such sales.
Grant revenues are generated primarily by our subsidiary Cell Cure Neurosciences Ltd (“Cell Cure”) from the Israel Innovation Authority (“IIA”) for the development of OpRegen® and our bio retina program, and from a Small Business Innovation Research grant from the National Institutes of Health for our vision restoration program (the “NIH grant”). The decreases in our grant revenues for the year ended December 31, 2020 as compared to the year ended December 31, 2019, were primarily due to less grant-related activities. Grant revenues generated by Cell Cure from the IIA for the development of OpRegen and our bio retina program (commencing in 2020) amounted to $0.7 million and $1.4 million for the years ended December 31, 2020 and 2019, respectively, and grant revenues generated by the NIH grant amounted to $0.4 million and $0.6 million for the years ended December 31, 2020 and 2019, respectively.
Royalties from product sales and license fees are generated from non-exclusive license agreements with multiple third parties. A majority of our royalties from product sales and license fees for the year ended December 31, 2020 are related to technologies that were acquired in the Asterias Merger. The decrease of $0.4 million for the year ended December 31, 2020 compared to the year ended December 31, 2019 was primarily related to the impact from a $0.6 million upfront, non-refundable payment for a new license agreement with a third party for the use of certain patents related to the culture of undifferentiated pluripotent stem cells in suspension that was recorded in 2019.
Operating Expenses
The following table shows our operating expenses for the years ended December 31, 2020 and 2019 (amounts in thousands, except percentages).
| Year Ended December 31, | $ | % | ||||||||||||||
| 2020 | 2019 | Decrease | Decrease | |||||||||||||
| Research and development expenses | $ | 12,317 | $ | 17,948 | $ | (5,631 | ) | (31 | %) | |||||||
| General and administrative expenses | 15,571 | (1) | 24,031 | (2) | (8,460 | ) | (35 | %) | ||||||||
| (1) | Includes $0.7 million of acquisition related costs for the Asterias Merger. |
| (2) | Includes $5.1 million of acquisition related costs for the Asterias Merger. |
| 60 |
Research and development expenses
Research and development expenses consist of costs incurred for company-sponsored, collaborative and contracted research and development activities. These costs include direct and research-related overhead expenses including compensation and related benefits, stock-based compensation, consulting fees, research and laboratory fees, rent of research facilities, amortization of intangible assets, and license fees paid to third parties to acquire patents or licenses to use patents and other technology. We expense research and development costs as incurred. Research and development expenses incurred and reimbursed by grants from third parties approximate the grant income recognized in the consolidated statements of operations.
The following table shows the amount of our total research and development expenses allocated to our primary research and development projects for the periods presented (amounts in thousands, except percentages).
Year Ended December 31, (unaudited) | ||||||||||||||||
| Amount | Percent of Total | |||||||||||||||
| Program | 2020 | 2019 | 2020 | 2019 | ||||||||||||
| OpRegen® and other ophthalmic applications | $ | 5,569 | $ | 12,069 | 45 | % | 67 | % | ||||||||
| OPC1 | 3,958 | 4,488 | 32 | % | 25 | % | ||||||||||
| VAC platform | 2,472 | 322 | 20 | % | 2 | % | ||||||||||
| Renevia and all other | 318 | 1,069 | 3 | % | 6 | % | ||||||||||
| Total research and development expenses | $ | 12,317 | $ | 17,948 | 100 | % | 100 | % | ||||||||
Research and development expenses for the year ended December 31, 2020 were $12.3 million as compared to $17.9 million for the year ended December 31, 2019. The decrease of $5.6 million is mainly attributable to the following:
| ● | a decrease of $6.5 million in OpRegen and other ophthalmic application expenses, attributable primarily to a decrease in manufacturing activities in 2020 as compared to 2019, | |
| ● | a decrease of $0.5 million in OPC1 related expenses, primarily related to return of unspent project funds of approximately $0.8 million from a former Asterias service provider, | |
| ● | a decrease of $0.8 million in Renevia and other related expenses as Renevia received a CE Mark in September 2019 and we are spending less on research activities as we are actively looking for a commercialization partner in Europe, offset by | |
| ● | an increase of $2.2 million in VAC program expenses, primarily related to the accrual of the signature fee of £1.25 million ($1.6 million) to Cancer Research UK. |
General and administrative expenses
General and administrative expenses include employee and director compensation allocated to general and administrative expenses, consulting fees other than those paid for science-related consulting, facilities and equipment rent and maintenance related expenses, insurance costs allocated to general and administrative expenses, costs of patent applications, prosecution and maintenance, stock exchange-related costs, depreciation expense, marketing costs, board fees, legal and accounting costs, and other miscellaneous expenses which are allocated to general and administrative expense.
General and administrative expenses for the year ended December 31, 2020 were $15.6 million as compared to $24.0 million for the year ended December 31, 2019. The total net decrease of $8.4 million was primarily attributable to a $5.5 million reduction in Asterias Merger related expenses, a $2.1 million reduction in compensation costs as a result of headcount reductions in 2019, a $0.9 million reduction in accounting expenses, a $0.5 million reduction in rent and utilities, a $0.3 million reduction in travel expenses, a $0.3 million reduction in office and information technology related expenses and a $0.2 million reduction in consulting expenses, offset by a $0.9 million increase related to the cessation of shared services reimbursements and a $0.5 million increase in legal and patent expenses.
| 61 |
Other income and expenses, net
The following table shows the amount of other income, net, during the year ended December 31, 2020 and 2019 (in thousands):
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Other income, net | ||||||||
| Interest income, net | $ | 1,039 | $ | 1,685 | ||||
| Gain on sale of marketable equity securities | 4,560 | 2,421 | ||||||
| Unrealized loss on marketable equity securities | (3,782 | ) | (2,898 | ) | ||||
| Gain on sale of equity method investment in OncoCyte | - | 546 | ||||||
| Unrealized gain on equity method investment in OncoCyte at fair value | - | 8,001 | ||||||
| Unrealized gain on equity method investment in Asterias at fair value | - | 6,744 | ||||||
| Unrealized (loss) gain on warrant liability | (174 | ) | 611 | |||||
| Other income, net | 2,880 | 2,532 | ||||||
| Total other income, net | $ | 4,523 | $ | 19,642 | ||||
Interest income and expense, net – During the years ended December 31, 2020 and 2019, we earned $1.0 million and $1.5 million of interest income, respectively, from our promissory note with Juvenescence Limited (“Juvenescence”).
Gain on equity method investment in Asterias – Prior to the closing of the Asterias Merger on March 8, 2019, we owned 21.7 million shares of common stock of Asterias, which we accounted for at fair value using the equity method of accounting. The fair value of our Asterias shares was approximately $20.2 million as of March 8, 2019, the closing date of the Asterias Merger, based on $0.93 per share, which was calculated by multiplying: (i) $1.31, the closing price of our common shares on such date; by (ii) the merger exchange ratio of 0.71. The fair value of our Asterias shares was approximately $13.5 million as of December 31, 2018, based on the closing price of Asterias common stock of $0.62 per share on such date. Accordingly, we recorded an unrealized gain of $6.7 million for the year ended December 31, 2019, representing the change in fair value of Asterias common stock from December 31, 2018 to March 8, 2019.
Gain (loss) on investment in OncoCyte – Prior to September 11, 2019, we elected to account for our shares of OncoCyte common stock at fair value using the equity method of accounting. We sold 2.25 million shares of OncoCyte common stock for net proceeds of $4.2 million in July 2019. Accordingly, our ownership in OncoCyte was reduced from 28% to 24%. We sold an additional 4.0 million shares of OncoCyte common stock for net proceeds of $6.5 million on September 11, 2019. Our ownership in OncoCyte was further reduced to 16% at this time. Effective September 11, 2019, we began accounting for our shares of OncoCyte common stock as marketable equity securities.
As of December 31, 2019, we had 8.4 million shares of OncoCyte common stock. These shares had a fair value of $19.0 million, based on the closing price of OncoCyte common stock of $2.25 per share on December 31, 2019.
As of December 31, 2020, we owned 3.6 million shares of OncoCyte common stock. These shares had a fair value of $8.7 million, based on the closing price of OncoCyte common stock of $2.39 per share on December 31, 2020,
For the year ended December 31, 2020, we recorded a realized gain of $3.1 million due to sales of OncoCyte shares in the period. In the same period, we also recorded an unrealized loss of $2.5 million related to our OncoCyte shares. The unrealized loss is comprised of $3.7 million related to the difference between the book cost basis of OncoCyte shares sold in the period versus the applicable prior month’s ending OncoCyte stock price, which is offset by $1.2 million related to the shares remaining at December 31, 2020 and the increase in OncoCyte’s stock price from $2.25 at December 31, 2019 to $2.39 at December 31, 2020. For the year ended December 31, 2019, we recorded a realized gain of $0.5 million due to sales of OncoCyte shares in the period. We also recorded an unrealized gain of $8.8 million due to the increase in OncoCyte’s stock price from $1.38 per share at December 31, 2018 to $2.25 per share at December 31, 2019. $8.0 million of the unrealized gain was recorded as an unrealized gain on an equity method investment as it was prior to September 11, 2019; the remaining $0.8 million was recorded as an unrealized gain on marketable equity securities.
| 62 |
All share prices are determined based on the closing price of OncoCyte common stock on the NYSE American on the applicable dates, or the last day of trading of the applicable quarter, if the last day of a quarter fell on a weekend.
We expect our other income and expenses, net, to continue to fluctuate each reporting period based on the changes in the market price of our OncoCyte shares, which could significantly impact our net income or loss reported in our condensed consolidated statements of operations for each period.
Marketable equity securities – We account for the shares we held in Hadasit Bio-Holdings (“HBL”) and AgeX as marketable equity securities, carried at fair market value on our consolidated balance sheets.
For the year ended December 31, 2020, we recorded realized gains of $0.8 million and $0.6 million due to sales of AgeX shares and HBL shares, respectively, in the period.
For the year ended December 31, 2020, we recorded unrealized losses of $1.3 million related to our AgeX shares. $0.5 million of the unrealized loss was related to the difference between the book cost basis of AgeX shares sold in the period versus the applicable prior month’s ending AgeX share price and an additional $0.8 million was related to mark to mark adjustments throughout the year on the remaining shares of AgeX at each applicable period.
Other income and expenses, net – Other income and expenses, net, in 2020 and 2019 consist primarily of net foreign currency transaction gains and losses recognized by Cell Cure and ESI, and changes in the fair value of the Cell Cure liability classified warrants. Foreign currency transaction gains and losses for the periods presented are principally related to the remeasurement of the U.S. dollar denominated notes payable by Cell Cure to Lineage.
Income Taxes
The market value of the shares of OncoCyte common stock we hold creates a deferred tax liability (the “OncoCyte DTL”) based on the closing prices of the shares, less our tax basis in the shares. The OncoCyte DTL is a source of future taxable income to us, as prescribed by ASC 740-10-30-17, that will more likely than not result in the realization of our deferred tax assets to the extent of the OncoCyte DTL. The OncoCyte DTL is determined based on the closing prices of the OncoCyte shares as of December 31, 2020. Due to the inherent unpredictability of future prices of those shares, we cannot reliably estimate or project the OncoCyte DTL on an annual basis. Therefore, the OncoCyte DTL is determined based on the actual closing prices on the last stock market trading day of the applicable accounting period, and the related impacts to the valuation allowance and deferred tax asset changes, and are recorded in the accounting period in which they occur.
In connection with the Asterias Merger, a deferred tax liability of $10.8 million (the “Asterias DTL”) was recorded as part of the acquisition accounting (see Note 3). The Asterias DTL is related to fair value adjustments for the assets and liabilities acquired in the Asterias Merger, principally consisting of IPR&D. This estimate of deferred taxes was determined based on the excess of the estimated fair values of the acquired assets and liabilities over the tax basis of the assets and liabilities acquired. The statutory tax rate was applied, as appropriate, to the adjustment based on the jurisdiction in which the adjustment is expected to occur. Because the IPR&D (prior to completion or abandonment of the R&D) is considered an indefinite-lived asset for accounting purposes, the fair value of the IPR&D on the acquisition date creates a deferred income tax liability in accordance with ASC 740. The Asterias DTL is computed using the fair value of the IPR&D assets on the acquisition date multiplied by Lineage’s respective federal and state income tax rates. While the Asterias DTL would reverse on impairment or sale or commencement of amortization of the related intangible assets, those events are not anticipated under ASC 740 for purposes of predicting reversal of a temporary difference to support the realization of deferred tax assets, except for certain deferred tax assets and credit carryforwards that are also indefinite in nature as of the Asterias Merger date, which may be considered for reversal under ASC 740 as further discussed below.
A valuation allowance is provided when it is more likely than not that some portion of the deferred tax assets will not be realized. Lineage established a full valuation allowance as of December 31, 2018 due to the uncertainty of realizing future tax benefits from its net operating loss carryforwards and other deferred tax assets, including foreign net operating losses generated by its subsidiaries. During the year ended December 31, 2019, a portion of the valuation allowance was released as it relates to Lineage’s indefinite lived assets that can be used against the indefinite lived liabilities. The amount of the valuation allowance released was $7.4 million; as new indefinite lived deferred tax assets are generated, we will continue to book provision benefits until the deferred tax liability position is exhausted, barring any new developments.
| 63 |
For the year ended December 31, 2020, Lineage recorded a $1.2 million deferred tax benefit for income taxes.
We expect that deferred income tax expense or benefit we record each reporting period, if any, will vary depending on the change in the closing stock prices of OncoCyte shares from period to period and the related changes in those deferred tax liabilities and our deferred tax assets and other credits, including changes in the valuation allowance, for each period.
See Note 3 to our consolidated financial statements included elsewhere in this Report for a description of the Asterias Merger that was completed on March 8, 2019. We have concluded that an ownership change did occur after the Asterias Merger, and the acquired operating loss carryforwards are subject to limitation under Section 382 of the Internal Revenue Service Code; Lineage will only be able to utilize $52.8 million of these operating loss carryforwards.
Liquidity and Capital Resources
At December 31, 2020, we had $41.6 million of cash, cash equivalents and marketable equity securities on hand, which includes our investments in OncoCyte and HBL. We may use our marketable equity securities for liquidity, as necessary, and as market conditions allow. The market value may not represent the amount that could be realized in a sale of investment shares due to various market and regulatory factors, including trading volume or market depth factors and volume and manner of sale restrictions under Federal securities laws, prevailing market conditions and prices at the time of any sale, and subsequent sales of securities by the entities. In addition, the value of our marketable equity securities may be significantly and adversely impacted by deteriorating global economic conditions and the recent disruptions to and volatility in the credit and financial markets in the United States and worldwide resulting from the ongoing COVID-19 pandemic.
Since inception, we have incurred significant operating losses and have funded our operations primarily through the issuance of equity securities, the sale of common stock of our former subsidiaries, AgeX and OncoCyte, payments from research grants, royalties from product sales and sales of research products and services. At December 31, 2020, we had an accumulated deficit of approximately $294.1 million, working capital of $36.2 million and shareholders’ equity of $95.1 million. We evaluated the projected cash flows for Lineage and our subsidiaries, and we believe that our $41.6 million in cash, cash equivalents and marketable equity securities at December 31, 2020, provide sufficient cash, cash equivalents, and liquidity to carry out our current planned operations through at least twelve months from the issuance date of our consolidated financial statements included elsewhere in this Report. If we need near term working capital or liquidity to supplement our cash and cash equivalents for our operations, we may sell some, or all, of our investments, as necessary.
On March 8, 2019, the Asterias Merger closed and Asterias became our wholly owned subsidiary. We began consolidating Asterias’ operations and results with our operations and results beginning on March 8, 2019. As we integrated Asterias’ operations into our own, we made extensive reductions in headcount and reduced non-clinical related spend, in each case, as compared to Asterias’ operations before the merger. We implemented significant cost savings initiatives and achieved reduced operational spend in 2020 compared to prior periods.
Our projected cash flows are subject to various risks and uncertainties, and the unavailability or inadequacy of financing to meet future capital needs could force us to modify, curtail, delay, or suspend some or all aspects of our current planned operations. Our determination as to when we will seek new financing and the amount of financing that we will need will be based on our evaluation of the progress we make in our research and development programs, any changes to the scope and focus of those programs, any changes in grant funding for certain of those programs, and projection of future costs, revenues, and rates of expenditure. Our ability to raise additional funds may be adversely impacted by deteriorating global economic conditions and the disruptions to and volatility in the credit and financial markets in the United States and worldwide resulting from the ongoing COVID-19 pandemic. We may be required to delay, postpone, or cancel our clinical trials or limit the number of clinical trial sites, unless we are able to obtain adequate financing. We cannot assure that adequate financing will be available on favorable terms, if at all. Sales of additional equity securities by us or our subsidiaries and affiliates could result in the dilution of the interests of our current shareholders.
| 64 |
Cash used in operating activities
Net cash used in operating activities of $19.8 million for the year ended December 31, 2020 primarily reflects the loss from operations of $26.4 million adjusted for the changes in assets and liabilities of $1.3 million. These items were offset primarily by non-cash expenses of $2.2 million for stock-based compensation and $2.1 million of depreciation and amortization. The unrealized gains on equity method investments and marketable securities, foreign currency remeasurement and deferred tax benefit are non-cash items that had no effect on cash flows.
Net cash used in operating activities of $31.9 million for the year ended December 31, 2019 primarily reflects the loss from operations of $38.9 million adjusted for the changes in assets and liabilities of $2.1 million. These items were offset primarily by non-cash expenses of $3.6 million for stock-based compensation and $3.1 million of depreciation and amortization. The unrealized gains on equity method investments and marketable securities, foreign currency remeasurement and deferred tax benefit are non-cash items that had no effect on cash flows.
Cash used in investing activities
Cash provided by investing activities of $13.0 million for the year ended December 31, 2020 was associated primarily with receipts of $10.9 million from sales of a portion of our OncoCyte holdings, $1.3 million in sales of our AgeX holdings and $0.8 million in sales of a portion of our HBL holdings.
Cash provided by investing activities of $17.0 million for the year ended December 31, 2019 was associated primarily with receipts of $10.7 million from sales of a portion of our OncoCyte holdings, $1.7 million in sales of a portion of our AgeX holdings and $1.7 million in sales of a portion of our HBL holdings as well as the receipt of $3.1 million of cash that Asterias had on the closing date of the Asterias Merger, offset by $0.4 million in purchases of equipment and other assets.
Cash provided by financing activities
Cash provided by financing activities of $29.9 million for the year ended December 31, 2020 was associated primarily with proceeds of $24.6 million from payment of the Juvenescence promissory note, gross proceeds of $5.1 million from sales of our common shares in at-the-market transactions under our Controlled Equity OfferingSM Sales Agreement with Cantor Fitzgerald & Co (which excludes $0.3 million of cash in transit related to 2020 sales that settled in 2021), and proceeds of $0.5 million from a Paycheck Protection Program (“PPP”) loan under the Coronavirus Aid, Relief, and Economic Security Act, all offset by $0.4 million in financing costs.
Cash provided by financing activities of $0.6 million for the year ended December 31, 2019 was associated primarily with $0.8 million in landlord reimbursements for tenant improvements, offset by $0.1 million in common shares received and retired for employee taxes paid.
Off-Balance Sheet Arrangements
As of December 31, 2020, we did not have any off-balance sheet arrangements, as defined under the rules of the Securities and Exchange Commission.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Under rules and regulations of the Securities and Exchange Commission, as a smaller reporting company, we are not required to provide the information required by this item.
| 65 |
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
Index to consolidated financial statements
See accompanying notes to consolidated financial statements.
| 66 |
Report of Independent Registered Public Accounting Firm
Shareholders and Board of Directors
Lineage Cell Therapeutics, Inc.
Carlsbad, California
Opinion on the Consolidated Financial Statements
We have audited the consolidated balance sheets of Lineage Cell Therapeutics, Inc. and Subsidiaries (collectively, the “Company”) as of December 31, 2020 and 2019, and the related consolidated statements of operations, comprehensive income (loss), changes in shareholders’ equity, and cash flows for each of the two years in the period ended December 31, 2020, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2020 and 2019, and the results of their operations and their cash flows for each of the two years in the period ended December 31, 2020, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the Audit Committee and that: (1) relates to accounts or disclosures that are material to the consolidated financial statements; and (2) involved our especially challenging, subjective, or complex judgments. The communication of a critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
| 67 |
Intangible Assets Impairment Assessment - In-Process Research and Development
Description of the Matter
As described in Note 7 to the consolidated financial statements, the Company’s in-process research and development indefinite-lived intangible assets had a balance of $46.5 million as of December 31, 2020.
Indefinite-lived intangible assets are tested for impairment annually and when events or changes in circumstances indicate that the asset might be impaired. As part of its indefinite-live intangible asset impairment assessment, management estimates the fair values of the Company’s indefinite-lived intangible assets using an income approach that utilizes a discounted cash flow model or, where appropriate, a market approach. The discounted cash flow model is dependent upon management’s estimates of future cash flows and other factors such as estimates of (i) future operating performance, including future sales, long-term growth rates, operating margins, discount rates, variations in the amount and timing of cash flows and the probability of achieving the estimated cash flows, and (ii) future economic conditions.
Auditing the Company’s impairment analysis of its indefinite-lived intangible assets is complex because of the significant judgment and estimates used by management in developing the fair value measurement of in-process research and development intangible assets. This in turn leads to significant audit effort and a high degree of auditor judgment and subjectivity in performing procedures to evaluate management’s estimated cash flows, including significant assumptions related to future sales, long-term growth rates, operating margins, discount rates, variations in the amount and timing of cash flows and the probability of achieving the estimated cash flows, and future economic conditions in determining the fair value of each of the in-process research and development intangible assets.
How We Addressed the Matter in Our Audit
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the consolidated financial statements. These procedures included, among others, obtaining an understanding of and evaluating management’s process for identifying potential impairment events; evaluating the appropriateness of the cash flow model used in the impairment testing process; testing the completeness, accuracy, and relevance of underlying data used in the model; and evaluating the reasonableness of the significant assumptions used by management, including the future cash flow projections and discount rates. We evaluated the reasonableness of management’s assumptions for future cash flow projections and discount rates in consideration of (i) the current and past performance of the asset group, (ii) the consistency with external market and industry data, and (iii) whether these assumptions were consistent with evidence obtained in other areas of the audit.
/s/ OUM & CO. LLP
San Francisco, California
March 11, 2021
We have served as the Company’s auditor since 2014.
| 68 |
LINEAGE CELL THERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
(IN THOUSANDS)
| December 31, 2020 | December 31, 2019 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS | ||||||||
| Cash and cash equivalents | $ | 32,585 | $ | 9,497 | ||||
| Marketable equity securities | 8,977 | 21,219 | ||||||
| Promissory note from Juvenescence (Note 5) | - | 23,616 | ||||||
| Trade accounts and grants receivable, net | 4 | 317 | ||||||
| Receivables from affiliates, net | - | 7 | ||||||
| Prepaid expenses and other current assets | 2,433 | 2,863 | ||||||
| Total current assets | 43,999 | 57,519 | ||||||
| NONCURRENT ASSETS | ||||||||
| Property and equipment, net (Notes 6 and 14) | 5,630 | 8,175 | ||||||
| Deposits and other long-term assets | 616 | 864 | ||||||
| Goodwill | 10,672 | 10,672 | ||||||
| Intangible assets, net | 47,032 | 48,248 | ||||||
| TOTAL ASSETS | $ | 107,949 | $ | 125,478 | ||||
| LIABILITIES AND SHAREHOLDERS’ EQUITY | ||||||||
| CURRENT LIABILITIES | ||||||||
| Accounts payable and accrued liabilities | $ | 6,813 | $ | 5,226 | ||||
| Financing lease and right-of-use liabilities, current portion (Note 14) | 762 | 1,223 | ||||||
| Deferred revenues | 193 | 45 | ||||||
| Liability classified warrants, current portion | 1 | - | ||||||
| Total current liabilities | 7,769 | 6,494 | ||||||
| LONG-TERM LIABILITIES | ||||||||
| Deferred tax liability | 2,076 | 3,315 | ||||||
| Deferred revenues, net of current portion | - | 200 | ||||||
| Right-of-use lease liability, net of current portion (Note 14) | 2,514 | 3,868 | ||||||
| Financing lease, net of current portion | 26 | 77 | ||||||
| Liability classified warrants and other long-term liabilities | 437 | 277 | ||||||
| TOTAL LIABILITIES | 12,822 | 14,231 | ||||||
| Commitments and contingencies (Note 14) | ||||||||
| SHAREHOLDERS’ EQUITY | ||||||||
| Preferred shares, no par value, authorized shares; issued and outstanding as of December 31, 2020 and 2019, respectively | - | - | ||||||
| Common shares, no par value, authorized shares; and shares issued and outstanding as of December 31, 2020 and 2019, respectively | 393,944 | 387,062 | ||||||
| Accumulated other comprehensive loss | (3,667 | ) | (681 | ) | ||||
| Accumulated deficit | (294,078 | ) | (273,422 | ) | ||||
| Lineage Cell Therapeutics, Inc. shareholders’ equity | 96,199 | 112,959 | ||||||
| Noncontrolling interest (deficit) | (1,072 | ) | (1,712 | ) | ||||
| Total shareholders’ equity | 95,127 | 111,247 | ||||||
| TOTAL LIABILITIES AND SHAREHOLDERS’ EQUITY | $ | 107,949 | $ | 125,478 | ||||
See accompanying notes to the consolidated financial statements.
| 69 |
LINEAGE CELL THERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS
(IN THOUSANDS, EXCEPT PER SHARE DATA)
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| REVENUES: | ||||||||
| Grant revenue | $ | 1,053 | $ | 2,037 | ||||
| Royalties from product sales and license fees | 773 | 1,221 | ||||||
| Sale of research products and services | - | 257 | ||||||
| Total revenues | 1,826 | 3,515 | ||||||
| Cost of sales | (385 | ) | (412 | ) | ||||
| Gross profit | 1,441 | 3,103 | ||||||
| OPERATING EXPENSES: | ||||||||
| Research and development | 12,317 | 17,948 | ||||||
| General and administrative | 15,571 | 24,031 | ||||||
| Total operating expenses | 27,888 | 41,979 | ||||||
| Loss from operations | (26,447 | ) | (38,876 | ) | ||||
| OTHER INCOME, NET: | ||||||||
| Interest income, net | 1,039 | 1,685 | ||||||
| Gain on sale of marketable securities | 4,560 | 2,421 | ||||||
| Gain on sale of equity method investment in OncoCyte | - | 546 | ||||||
| Unrealized loss on marketable equity securities | (3,782 | ) | (2,898 | ) | ||||
| Unrealized gain on equity method investment in OncoCyte at fair value | - | 8,001 | ||||||
| Unrealized gain on equity method investment in Asterias at fair value | - | 6,744 | ||||||
| Unrealized (loss) gain on warrant liability | (174 | ) | 611 | |||||
| Other income, net | 2,880 | 2,532 | ||||||
| Total other income, net | 4,523 | 19,642 | ||||||
| LOSS BEFORE INCOME TAXES | (21,924 | ) | (19,234 | ) | ||||
| Income tax benefit | 1,239 | 7,407 | ||||||
| NET LOSS | (20,685 | ) | (11,827 | ) | ||||
| Net loss attributable to noncontrolling interest | 36 | 118 | ||||||
| NET LOSS ATTRIBUTABLE TO LINEAGE | $ | (20,649 | ) | $ | (11,709 | ) | ||
| NET LOSS PER COMMON SHARE: | ||||||||
| BASIC AND DILUTED | $ | (0.14 | ) | $ | (0.08 | ) | ||
| WEIGHTED AVERAGE NUMBER OF COMMON SHARES OUTSTANDING: | ||||||||
| BASIC AND DILUTED | 150,044 | 145,533 | ||||||
See accompanying notes to the consolidated financial statements.
| 70 |
LINEAGE CELL THERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(IN THOUSANDS)
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| NET LOSS | $ | (20,685 | ) | $ | (11,827 | ) | ||
| Other comprehensive loss, net of tax: | ||||||||
| Foreign currency translation adjustments, net of tax | (2,986 | ) | (2,107 | ) | ||||
| COMPREHENSIVE LOSS | (23,671 | ) | (13,934 | ) | ||||
| Less: comprehensive loss attributable to noncontrolling interest | 36 | 118 | ||||||
| COMPREHENSIVE LOSS ATTRIBUTABLE TO LINEAGE COMMON SHAREHOLDERS | $ | (23,635 | ) | $ | (13,816 | ) | ||
See accompanying notes to the consolidated financial statements.
| 71 |
LINEAGE CELL THERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN SHAREHOLDERS’ EQUITY
(IN THOUSANDS)
| Preferred Shares | Common Shares | Accumulated | ||||||||||||||||||||||||||||||
Number of Shares | Amount | Number of Shares | Amount | Accumulated Deficit | Noncontrolling Interest/(Deficit) | Other Comprehensive Income/(Loss) | Total Shareholders’ Equity | |||||||||||||||||||||||||
| BALANCE AT DECEMBER 31, 2018 | - | $ | - | 127,136 | $ | 354,270 | $ | (261,856 | ) | $ | (1,594 | ) | $ | 1,426 | $ | 92,246 | ||||||||||||||||
| Shares issued in connection with the Asterias Merger | - | 24,696 | 32,352 | - | - | - | 32,352 | |||||||||||||||||||||||||
| Shares retired in connection with the Asterias Merger | - | (2,622 | ) | (3,435 | ) | - | - | - | (3,435 | ) | ||||||||||||||||||||||
| Shares issued for settlement of Lineage Warrants | - | 252 | 302 | - | - | - | 302 | |||||||||||||||||||||||||
| Shares issued upon vesting of restricted stock units, net of shares retired to pay employees’ taxes | - | 189 | (110 | ) | - | - | - | (110 | ) | |||||||||||||||||||||||
| Stock-based compensation | - | 3,501 | - | - | - | 3,501 | ||||||||||||||||||||||||||
| Stock-based compensation for shares issued upon vesting of Asterias restricted stock units attributable to post combination services | - | 60 | 79 | - | - | - | 79 | |||||||||||||||||||||||||
| Shares issued through ATM | - | 93 | 103 | - | - | - | 103 | |||||||||||||||||||||||||
| Adjustment upon adoption of leasing standard | - | - | - | - | 143 | - | - | 143 | ||||||||||||||||||||||||
| Foreign currency translation gain (loss) | - | - | - | - | - | - | (2,107 | ) | (2,107 | ) | ||||||||||||||||||||||
| NET LOSS | - | - | - | - | (11,709 | ) | (118 | ) | - | (11,827 | ) | |||||||||||||||||||||
| BALANCE AT DECEMBER 31, 2019 | $ | 149,804 | $ | 387,062 | $ | (273,422 | ) | $ | (1,712 | ) | $ | (681 | ) | $ | 111,247 | |||||||||||||||||
| Shares issued through ATM | - | 3,095 | 5,404 | - | - | - | 5,404 | |||||||||||||||||||||||||
| Shares issued upon vesting of restricted stock units, net of shares retired to pay employees’ taxes | - | 47 | (27 | ) | - | - | - | (27 | ) | |||||||||||||||||||||||
| Shares issued for services | - | 150 | 119 | - | - | - | 119 | |||||||||||||||||||||||||
| Stock-based compensation | - | 2,227 | - | - | - | 2,227 | ||||||||||||||||||||||||||
| Financing related fees | - | - | - | (209 | ) | - | - | - | (209 | ) | ||||||||||||||||||||||
| Dissolution of BioTime Asia | - | - | - | (676 | ) | (7 | ) | 676 | - | (7 | ) | |||||||||||||||||||||
| Hadasit non-cash warrant exercise | - | - | - | 44 | - | - | - | 44 | ||||||||||||||||||||||||
| Foreign currency translation gain (loss) | - | - | - | - | - | - | (2,986 | ) | (2,986 | ) | ||||||||||||||||||||||
| NET LOSS | - | - | - | - | (20,649 | ) | (36 | ) | - | (20,685 | ) | |||||||||||||||||||||
| BALANCE AT DECEMBER 31, 2020 | $ | 153,096 | $ | 393,944 | $ | (294,078 | ) | $ | (1,072 | ) | $ | (3,667 | ) | $ | 95,127 | |||||||||||||||||
See accompanying notes to the consolidated financial statements.
| 72 |
LINEAGE CELL THERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
(IN THOUSANDS)
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss attributable to Lineage | $ | (20,649 | ) | $ | (11,709 | ) | ||
| Net loss attributable to noncontrolling interest | (36 | ) | (118 | ) | ||||
| Adjustments to reconcile net loss attributable to Lineage to net cash used in operating activities: | ||||||||
| Unrealized gain on equity method investment in OncoCyte at fair value | - | (8,001 | ) | |||||
| Unrealized gain on equity method investment in Asterias at fair value | - | (6,744 | ) | |||||
| Gain on sale of marketable equity securities | (4,560 | ) | (2,967 | ) | ||||
| Unrealized loss on marketable equity securities | 3,782 | 2,898 | ||||||
| Income tax benefit | (1,239 | ) | (7,407 | ) | ||||
| Depreciation expense, including amortization of leasehold improvements | 823 | 1,002 | ||||||
| Amortization of right-of-use assets | 72 | 129 | ||||||
| Amortization of intangible assets | 1,216 | 1,998 | ||||||
| Stock-based compensation | 2,227 | 3,580 | ||||||
| Common stock issued for services | 119 | - | ||||||
| Change in unrealized loss (gain) on warrant liability | 174 | (611 | ) | |||||
| Write-off of security deposit | 150 | - | ||||||
| Amortization of deferred license fee | (200 | ) | - | |||||
| Foreign currency remeasurement and other (gain) loss | (2,957 | ) | (2,367 | ) | ||||
| (Gain) loss on sale of assets | (20 | ) | 273 | |||||
| Realized loss on warrant exercise | 44 | - | ||||||
| Dividend received | - | 182 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts and grants receivable, net | 287 | 467 | ||||||
| Accrued interest receivable | (1,008 | ) | (1,512 | ) | ||||
| Receivables from affiliates, net of payables | 7 | 2,105 | ||||||
| Prepaid expenses and other current assets | 1,575 | (260 | ) | |||||
| Accounts payable and accrued liabilities | 308 | (2,885 | ) | |||||
| Deferred revenue and other liabilities | 132 | - | ||||||
| Net cash used in operating activities | (19,753 | ) | (31,947 | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||
| Proceeds from sale of OncoCyte common shares | 10,941 | 10,738 | ||||||
| Proceeds from the sale of AgeX common shares | 1,290 | 1,734 | ||||||
| Proceeds from the sale of Hadasit common shares | 830 | 1,743 | ||||||
| Cash and cash equivalents acquired in the Asterias Merger | - | 3,117 | ||||||
| Purchase of property and equipment | (64 | ) | (440 | ) | ||||
| Proceeds from sale of assets | 23 | 82 | ||||||
| Security deposit paid and other | 18 | (17 | ) | |||||
| Net cash provided by investing activities | 13,038 | 16,957 | ||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Proceeds from payment of Juvenescence promissory note | 24,624 | - | ||||||
| Common shares received and retired for employee taxes paid | (27 | ) | (110 | ) | ||||
| Proceeds from sale of subsidiary warrants | - | (40 | ) | |||||
| Proceeds from sale of common shares | 5,127 | 103 | ||||||
| Payments for offering costs | (356 | ) | - | |||||
| Repayment of financing lease liabilities | (26 | ) | (30 | ) | ||||
| Proceeds from Paycheck Protection Program (“PPP”) Loan (Note 8) | 523 | - | ||||||
| Reimbursement from landlord on tenant improvements | - | 764 | ||||||
| Repayment of principal portion of promissory notes | - | (70 | ) | |||||
| Net cash provided by financing activities | 29,865 | 617 | ||||||
| Effect of exchange rate changes on cash, cash equivalents and restricted cash | (63 | ) | 70 | |||||
| NET INCREASE (DECREASE) IN CASH, CASH EQUIVALENTS AND RESTRICTED CASH | 23,087 | (14,303 | ) | |||||
| At beginning of year | 10,096 | 24,399 | ||||||
| At end of year | $ | 33,183 | $ | 10,096 | ||||
| SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION: | ||||||||
| Cash paid during year for interest | $ | 20 | $ | 28 | ||||
| SUPPLEMENTAL SCHEDULE OF NON-CASH FINANCING AND INVESTING ACTIVITIES: | ||||||||
| Receivable from sale of common shares in at the market offering | $ | 269 | $ | |||||
| Receivable from sale of AgeX common shares | - | 41 | ||||||
| Issuance of common shares for the Asterias Merger (Note 3) | - | 32,353 | ||||||
| Assumption of liabilities in the Asterias Merger | - | 982 | ||||||
| Assumption of warrants in the Asterias Merger | - | 867 | ||||||
| Issuance of common shares for settlement of Lineage Warrants | - | 332 | ||||||
See accompanying notes to the consolidated financial statements.
| 73 |
LINEAGE CELL THERAPEUTICS, INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
1. Organization, Basis of Presentation and Liquidity
General – Lineage Cell Therapeutics, Inc. (“Lineage”) is a clinical-stage biotechnology company developing novel cell therapies for unmet medical needs. Lineage’s focus is to develop therapies for degenerative retinal diseases, neurological conditions associated with demyelination, and aiding the body in detecting and combating cancer. Specifically, Lineage is testing therapies to treat dry age-related macular degeneration, spinal cord injuries, and non-small cell lung cancer. Lineage’s programs are based on its proprietary cell-based therapy platform and associated development and manufacturing capabilities. From this platform, Lineage develops and manufactures specialized, terminally or functionally differentiated human cells from its pluripotent and progenitor cell starting materials. These differentiated cells are transplanted into a patient either to replace or support cells that are dysfunctional or absent due to degenerative disease or traumatic injury, or administered as a means of helping the body mount an effective immune response to cancer.
Lineage has allogeneic, or “off-the-shelf,” cell therapy programs in clinical development:
| ● | OpRegen®, a retinal pigment epithelium cell replacement therapy currently in a Phase 1/2a multicenter clinical trial for the treatment of advanced dry age-related macular degeneration (“AMD”) with geographic atrophy. There currently are no therapies approved by the U.S. Food and Drug Administration (“FDA”) for dry AMD, which accounts for approximately 85-90% of all AMD cases and is the leading cause of blindness in people over the age of 60. | |
| ● | OPC1, an oligodendrocyte progenitor cell therapy currently in a Phase 1/2a multicenter clinical trial for acute spinal cord injuries (“SCI”). This clinical trial has been partially funded by the California Institute for Regenerative Medicine. | |
| ● | VAC2, cancer immunotherapy of antigen-presenting dendritic cells currently in a Phase 1 clinical trial in non-small cell lung cancer. This clinical trial is being funded and conducted by Cancer Research UK, the world’s largest independent cancer research charity. |
In addition to seeking to create value for shareholders by developing product candidates and other technologies through our clinical development programs, we also seek to create value from our technologies through partnering and strategic transactions. We founded two companies that later became publicly traded companies: OncoCyte Corporation (“OncoCyte”) and AgeX Therapeutics, Inc. (“AgeX”).
During the year ended December 31, 2020, we received approximately $12.6 million in gross proceeds in connection with our sale of shares of OncoCyte and AgeX. In August 2020, we also received $24.6 million from Juvenescence Limited (“Juvenescence”), representing principal and accrued interest under a promissory note we received in connection with our sale of AgeX shares to Juvenescence in August 2018.
We no longer hold any common stock in AgeX. The value of our OncoCyte holdings as of March 5, 2021, was approximately $4.2 million, based on the closing price of its common stock on that date.
Though our principal focus is on advancing our three cell therapy programs currently in clinical development, we may seek to create additional value through corporate transactions, as we have in the past, or by initiating new programs using our protocols or with new protocols and cell lines.
Asterias Merger
On November 7, 2018, Lineage, Asterias Biotherapeutics, Inc. (“Asterias”) and Patrick Merger Sub, Inc., a wholly owned subsidiary of Lineage, entered into an Agreement and Plan of Merger (the “Merger Agreement”) whereby Lineage agreed to acquire all of the outstanding common stock of Asterias in a stock-for-stock transaction (the “Asterias Merger”).
On March 7, 2019, the shareholders of each of Lineage and Asterias approved the Merger Agreement. Prior to the Asterias Merger, Lineage owned approximately 38% of Asterias’ issued and outstanding common stock and accounted for Asterias as an equity method investment.
| 74 |
On March 8, 2019, the Asterias Merger closed with Asterias surviving as a wholly owned subsidiary of Lineage. The former stockholders of Asterias (other than Lineage) received common shares of Lineage for every share of Asterias common stock they owned. Lineage issued common shares, including shares issued in respect of restricted stock units issued by Asterias that immediately vested in connection with the closing of the Asterias Merger. The aggregate dollar value of such shares, based on the closing price of Lineage common shares on March 8, 2019, was $32.4 million. Lineage also assumed warrants to purchase shares of Asterias common stock.
The Asterias Merger has been accounted for using the acquisition method of accounting in accordance with Accounting Standards Codification (“ASC”) Topic 805, Business Combinations, which requires, among other things, that the assets and liabilities assumed be recognized at their fair values as of the acquisition date.
See Note 3 for a full discussion of the Asterias Merger.
Investment in OncoCyte
Lineage has significant equity holdings in OncoCyte, which Lineage founded and, in the past, was a majority-owned consolidated subsidiary until February 17, 2017, when Lineage deconsolidated OncoCyte’s financial statements. OncoCyte is focused on developing and commercializing laboratory-developed tests to serve unmet medical needs across the cancer care continuum. As of December 31, 2020, Lineage owned approximately million shares of OncoCyte common stock, or 5.4% of its outstanding shares (see Note 4).
Use of estimates
The preparation of consolidated financial statements in conformity with accounting principles generally accepted in the U.S. (“GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period with consideration given to materiality. Significant estimates and assumptions which are subject to significant judgment include those related to going concern assessment of consolidated financial statements, useful lives associated with long-lived assets, including evaluation of asset impairment, allowances for uncollectible accounts receivables, loss contingencies, deferred income taxes and tax reserves, including valuation allowances related to deferred income taxes, and assumptions used to value stock-based awards, debt or other equity instruments. Actual results could differ materially from those estimates.
Principles of consolidation
Lineage’s consolidated financial statements include the accounts of its subsidiaries. The following table reflects Lineage’s ownership, directly or through one or more subsidiaries, of the outstanding shares of its operating subsidiaries as of December 31, 2020.
| Subsidiary | Field of Business | Lineage Ownership | Country | |||||
| Asterias BioTherapeutics, Inc. | Cell therapy clinical development programs in spinal cord injury and oncology | 100 | % | USA | ||||
| Cell Cure Neurosciences Ltd (“Cell Cure”) | Development and manufacturing of Lineage’s cell replacement platform technology | 99 | %(1) | Israel | ||||
| ES Cell International Pte. Ltd. (“ESI”) | Stem cell products for research, including clinical grade cell lines produced under cGMP | 100 | % | Singapore | ||||
| OrthoCyte Corporation (“OrthoCyte”) | Developing bone grafting products for orthopedic diseases and injuries | 99.8 | % | USA | ||||
| (1) |
All material intercompany accounts and transactions have been eliminated in consolidation. As of December 31, 2020, Lineage consolidated its direct and indirect wholly owned or majority-owned subsidiaries because Lineage has the ability to control their operating and financial decisions and policies through its ownership, and the noncontrolling interest is reflected as a separate element of shareholders’ equity on Lineage’s consolidated balance sheets.
| 75 |
Liquidity
Since inception, Lineage has incurred significant operating losses and has funded its operations primarily through sale of common stock of AgeX and OncoCyte, both former subsidiaries, sale of common stock of Hadasit Bio-Holdings (“HBL”), receipt of research grants, royalties from product sales, license revenues, sales of research products and issuance of equity securities.
On May 1, 2020, Lineage entered into a Controlled Equity OfferingSM Sales Agreement (the “Sales Agreement”) with Cantor Fitzgerald & Co., as sales agent (“Cantor Fitzgerald”), pursuant to which Lineage may, but is not obligated to, raise up to $million through the sale of common shares (“ATM Shares”) from time to time in at-the-market transactions under the Sales Agreement. As of December 31, 2020, Lineage raised $5.1 million in gross proceeds under the Sales Agreement (which excludes $0.3 million in cash in transit related to 2020 sales that settled in 2021) and during the first quarter through March 5, 2021, Lineage raised $19.9 million in gross proceeds under the Sales Agreement (which includes $0.3 million in cash in transit related to 2020 sales that settled in 2021). On March 5, 2021, Lineage filed a prospectus supplement with the Securities and Exchange Commission (the “SEC”) in connection with the offer and sale of an additional $25 million of ATM Shares.
At December 31, 2020, Lineage had an accumulated deficit of approximately $294.1 million, working capital of $36.2 million and shareholders’ equity of $95.1 million. Lineage has evaluated its projected cash flows and believes that its $41.6 million of cash, cash equivalents and marketable equity securities are sufficient to fund Lineage’s planned operations for at least the next twelve months from the issuance date of the condensed consolidated financial statements included herein. If Lineage needs near term working capital or liquidity to supplement its cash and cash equivalents for its operations, Lineage may sell some, or all, of its marketable equity securities, as necessary.
On March 8, 2019, Asterias became Lineage’s wholly owned subsidiary, and Lineage began consolidating Asterias’ operations and results with its operations and results (see Note 3). Lineage has made extensive reductions in headcount and reduced non-clinical related spend, in each case, as compared to Asterias’ operations before the Asterias Merger.
Lineage’s projected cash flows are subject to various risks and uncertainties, and the unavailability or inadequacy of financing to meet future capital needs could force Lineage to modify, curtail, delay, or suspend some or all aspects of its planned operations. Lineage’s determination as to when it will seek new financing and the amount of financing that it will need will be based on Lineage’s evaluation of the progress it makes in its research and development programs, any changes to the scope and focus of those programs, any changes in grant funding for certain of those programs, and projection of future costs, revenues, and rates of expenditure. Lineage’s ability to raise additional funds may be adversely impacted by deteriorating global economic conditions and the disruptions to and volatility in the credit and financial markets in the United States and worldwide resulting from the ongoing COVID-19 pandemic. Lineage may be required to delay, postpone, or cancel clinical trials or limit the number of clinical trial sites, unless it is able to obtain adequate financing. In addition, Lineage has incurred significant costs in connection with the acquisition of Asterias and with integrating its operations. Lineage may incur additional costs to maintain employee morale and to retain key employees. Lineage cannot assure that adequate financing will be available on favorable terms, if at all. Sales of additional equity securities by Lineage or its subsidiaries and affiliates could result in the dilution of the interests of current shareholders.
2. Summary of Significant Accounting Policies
Business Combinations – Lineage accounts for business combinations, such as the Asterias Merger completed in March 2019, in accordance with ASC Topic 805, which requires the purchase price to be measured at fair value. When the purchase consideration consists entirely of Lineage common shares, Lineage calculates the purchase price by determining the fair value, as of the acquisition date, of shares issued in connection with the closing of the acquisition. Lineage recognizes estimated fair values of the tangible assets and intangible assets acquired, including in-process research and development (“IPR&D”), and liabilities assumed as of the acquisition date, and records as goodwill any amount of the fair value of the tangible and intangible assets acquired and liabilities assumed in excess of the purchase price.
Marketable Equity Securities – Lineage accounts for the shares it holds in OncoCyte and HBL (and AgeX previously) as marketable equity securities in accordance with ASC 320-10-25, Investments – Debt and Equity Securities, as amended by Accounting Standards Update (“ASU”) 2016-01, Financial Instruments–Overall: Recognition and Measurement of Financial Assets and Financial Liabilities, further discussed below.
| 76 |
The OncoCyte and AgeX shares have readily determinable fair values quoted on the NYSE American under trading symbols “OCX” and “AGE”. The HBL shares have a readily determinable fair value quoted on the Tel Aviv Stock Exchange (“TASE”) under trading symbol “HDST” where share prices are denominated in New Israeli Shekels (NIS).
Prior to September 11, 2019, Lineage accounted for its OncoCyte shares held at fair value, using the equity method of accounting. On September 11, 2019, Lineage’s ownership percentage decreased from 24% to 16% when it sold million shares of OncoCyte common stock. Accordingly, as the ownership percentage was reduced to less than 20%, Lineage is no longer considered to exercise significant influence over OncoCyte and is now accounting for its OncoCyte holdings as marketable equity securities. Prior to the Asterias Merger completed on March 8, 2019, Lineage accounted for its Asterias shares held at fair value, using the equity method of accounting.
Revenue Recognition – Lineage recognizes revenue in accordance with Financial Accounting Standards Board (“FASB”) Accounting Standards Update (“ASU”) ASU 2014-09, Revenues from Contracts with Customers (Topic 606), and in a manner that depicts the transfer of control of a product or a service to a customer and reflects the amount of the consideration it is entitled to receive in exchange for such product or service. In doing so, Lineage follows a five-step approach: (i) identify the contract with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations; and (v) recognize revenue when (or as) the customer obtains control of the product or service. Lineage considers the terms of a contract and all relevant facts and circumstances when applying the revenue recognition standard. Lineage applies the revenue recognition standard, including the use of any practical expedients, consistently to contracts with similar characteristics and in similar circumstances.
Lineage’s largest source of revenue is currently related to government grants. In applying the provisions of ASU 2014-09, Lineage has determined that government grants are out of the scope of ASU 2014-09 because the government entities do not meet the definition of a “customer,” as defined by ASU 2014-09, as there is not considered to be a transfer of control of good or services to the government entities funding the grant. Lineage has, and will continue to, account for grants received to perform research and development services in accordance with ASC 730-20, Research and Development Arrangements, which requires an assessment, at the inception of the grant, of whether the grant is a liability or a contract to perform research and development services for others. If Lineage or a subsidiary receiving the grant is obligated to repay the grant funds to the grantor regardless of the outcome of the research and development activities, then Lineage is required to estimate and recognize that liability. Alternatively, if Lineage or a subsidiary receiving the grant is not required to repay, or if it is required to repay the grant funds only if the research and development activities are successful, then the grant agreement is accounted for as a contract to perform research and development services for others, in which case, grant revenue is recognized when the related research and development expenses are incurred (see Note 14).
Deferred grant revenues represent grant funds received from the governmental funding agencies for which the allowable expenses have not yet been incurred as of the balance sheet date reported. As of December 31, 2020, deferred grant revenue was $193,000.
For the years ended December 31, 2020 and 2019, respectively, Lineage reported a net loss attributable to common shareholders, and therefore, all potentially dilutive common shares were considered antidilutive for those periods.
| 77 |
Years Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Stock options | 16,215 | 15,060 | ||||||
| Lineage Warrants (1) (Note 3) | 1,090 | 1,090 | ||||||
| Restricted stock units | 93 | 166 | ||||||
| (1) |
Restricted Cash – In accordance with ASU 2016-18, Statement of Cash Flows (Topic 230): Restricted Cash, Lineage explains the change during the year in the total of cash, cash equivalents and restricted cash, and includes restricted cash with cash and cash equivalents when reconciling the beginning-of-year and end-of-year total amounts shown on the condensed consolidated statements of cash flows.
Lineage has several certificates of deposit as required under our facility leases and credit card program. Lineage is restricted from using this cash for working capital purposes. At December 31, 2020, Lineage maintains $420,000 pursuant to the Cell Cure Leases, $100,000 pursuant to its credit card program and $78,000 pursuant to the Alameda Lease. Amounts related to the Cell Cure Leases and credit card program are recorded in deposits and other long-term assets and the amount related to the Alameda Lease is recorded in prepaid expenses and other current assets, as this certificate of deposit is expected to be released within the first quarter of 2021.
The following table provides a reconciliation of cash, cash equivalents, and restricted cash reported within the condensed consolidated balance sheet dates that comprise the total of the same such amounts shown in the condensed consolidated statements of cash flows for all periods presented herein (in thousands):
December 31, 2020 | December 31, 2019 | |||||||
| Cash and cash equivalents | $ | 32,585 | $ | 9,497 | ||||
| Restricted cash included in deposits and other long-term assets (see Note 14) | 520 | 599 | ||||||
| Restricted cash included in prepaid expenses and other current assets (see Note 14) | 78 | |||||||
| Total cash, cash equivalents, and restricted cash as shown in the condensed consolidated statements of cash flows | $ | 33,183 | $ | 10,096 | ||||
Lease accounting and impact of adoption of the new lease standard – On January 1, 2019, Lineage adopted ASU 2016-02, Leases (Topic 842, “ASC 842”) and its subsequent amendments affecting Lineage: (i) ASU 2018-10, Codification Improvements to Topic 842, Leases; and (ii) ASU 2018-11, Leases (Topic 842): Targeted improvements, using the modified retrospective method.
Lineage management determines if an arrangement is a lease at inception. Leases are classified as either financing or operating, with classification affecting the pattern of expense recognition in the consolidated statements of operations. When determining whether a lease is a finance lease or an operating lease, ASC 842 does not specifically define criteria to determine “major part of remaining economic life of the underlying asset” and “substantially all of the fair value of the underlying asset.” For lease classification determination, Lineage continues to use: (i) greater than or equal to 75% to determine whether the lease term is a major part of the remaining economic life of the underlying asset; and (ii) greater than or equal to 90% to determine whether the present value of the sum of lease payments is substantially all of the fair value of the underlying asset. Under the available practical expedients, Lineage accounts for the lease and non-lease components as a single lease component. Lineage recognizes right-of-use (“ROU”) assets and lease liabilities for leases with terms greater than twelve months in the condensed consolidated balance sheet.
| 78 |
ROU assets represent Lineage’s right to use an underlying asset during the lease term and lease liabilities represent Lineage’s obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized at commencement date based on the present value of lease payments over the lease term. As most of Lineage’s leases do not provide an implicit rate, Lineage uses its incremental borrowing rate based on the information available at commencement date in determining the present value of lease payments. Lineage uses the implicit rate when readily determinable. The operating lease ROU asset also includes any lease payments made and excludes lease incentives. Lineage’s lease terms may include options to extend or terminate the lease when it is reasonably certain that Lineage will exercise that option. Lease expense for lease payments is recognized on a straight-line basis over the lease term.
Operating leases are included as right-of-use assets in property and equipment (see Note 6), and ROU lease liabilities, current and long-term, in the condensed consolidated balance sheets. Financing leases are included in property and equipment, and in financing lease liabilities, current and long-term, in Lineage’s condensed consolidated balance sheets.
In connection with the adoption on ASC 842 on January 1, 2019, Lineage derecognized net book value of leasehold improvements and corresponding lease liabilities of $1.9 million and $2.0 million, respectively, which was the carrying value of certain operating leases as of December 31, 2018, included in property and equipment and lease liabilities, respectively, recorded pursuant to build to suit lease accounting under the previous ASC 840 lease standard. The derecognition of these amounts from the superseded ASC 840 lease standard was offset by a cumulative effect adjustment of $0.1 million as a reduction of Lineage’s accumulated deficit on January 1, 2019. These build to suit leases were primarily related to Lineage’s prior leases in Alameda, California and Cell Cure’s leases in Jerusalem, Israel (See Note 14). ASC 842 requires build to suit leases recognized on Lineage’s consolidated balance sheets as of December 31, 2018 to be derecognized upon the adoption of the new lease standard and be recognized in accordance with the new standard on January 1, 2019.
The adoption of ASC 842 had a material impact in Lineage’s consolidated balance sheets, with the most significant impact resulting from the recognition of ROU assets and lease liabilities for operating leases with remaining terms greater than twelve months on the adoption date. Lineage’s accounting for financing leases (previously referred to as “capital leases”) remained substantially unchanged (see Note 14).
Goodwill and IPR&D – Goodwill is calculated as the difference between the acquisition date fair value of the consideration transferred and the values assigned to the assets acquired and liabilities assumed. Goodwill is not amortized but is tested for impairment at least annually, or more frequently if circumstances indicate potential impairment. IPR&D assets are indefinite-lived intangible assets until the completion or abandonment of the associated research and development (“R&D”) efforts. Once the R&D efforts are completed or abandoned, the IPR&D will either be amortized over the asset life as a finite-lived intangible asset or be impaired, respectively, in accordance with ASC 350, Intangibles – Goodwill and Other. In accordance with ASC 350, goodwill and acquired IPR&D are determined to have indefinite lives and, therefore, are not amortized. Instead, they are tested for impairment at least annually and between annual tests if Lineage becomes aware of an event or a change in circumstances that would indicate the asset may be impaired.
Going concern assessment – Lineage assesses going concern uncertainty for its consolidated financial statements to determine if Lineage has sufficient cash and cash equivalents on hand and working capital to operate for a period of at least one year from the date the consolidated financial statements are issued or are available to be issued, which is referred to as the “look-forward period” as defined by FASB’s ASU No. 2014-15. As part of this assessment, based on conditions that are known and reasonably knowable to Lineage, Lineage will consider various scenarios, forecasts, projections, and estimates, and Lineage will make certain key assumptions, including the timing and nature of projected cash expenditures or programs, and its ability to delay or curtail those expenditures or programs, if necessary, among other factors. Based on this assessment, as necessary or applicable, Lineage makes certain assumptions concerning its ability to curtail or delay research and development programs and expenditures within the look-forward period in accordance with ASU No. 2014-15.
| 79 |
Cash and cash equivalents – Lineage considers all highly liquid investments purchased with an original maturity of three months or less to be cash equivalents. As of December 31, 2020 and 2019, Lineage had $28.8 million and $6.6 million in money market funds, respectively, considered to be cash equivalents.
Concentrations of credit risk and significant sources of supply – Financial instruments that potentially subject Lineage to significant concentrations of credit risk consist primarily of cash and cash equivalents. Lineage limits the amount of credit exposure of cash balances by maintaining its accounts in high credit quality financial institutions. Cash equivalent deposits with financial institutions may occasionally exceed the limits of insurance on bank deposits; however, Lineage has not experienced any losses on such accounts.
Lineage relies on single-source, third-party suppliers for a few key components of our product candidates. If these single-source, third-party suppliers are unable to continue providing a key component, the initiation or progress of any clinical studies of its product candidates may be impeded.
Property and equipment, net – Property and equipment is stated at cost and is being depreciated using the straight-line method over their estimated useful lives ranging from 3 to 10 years. Leasehold improvements are amortized over the shorter of the useful life or the lease term. (See Note 6).
Long-lived intangible assets – Long-lived intangible assets, consisting primarily of acquired patents, patent applications, and licenses to use certain patents are stated at acquired cost, less accumulated amortization. Amortization expense is computed using the straight-line method over the estimated useful lives of the assets, generally over 5 to 10 years.
Impairment of long-lived assets – Long-lived assets, including long-lived intangible assets, are reviewed annually for impairment and whenever events or changes in circumstances indicate that the carrying amount of an asset may not be fully recoverable. If an impairment indicator is present, Lineage evaluates recoverability by a comparison of the carrying amount of the assets to future undiscounted net cash flows expected to be generated by the assets. If the assets are impaired, the impairment recognized is measured by the amount by which the carrying amount exceeds the estimated fair value of the assets.
Accounting for warrants – Lineage determines the accounting classification of warrants that it or its subsidiaries issue, as either liability or equity, by first assessing whether the warrants meet liability classification in accordance with ASC 480-10, Accounting for Certain Financial Instruments with Characteristics of both Liabilities and Equity, and then in accordance with ASC 815-40, Accounting for Derivative Financial Instruments Indexed to, and Potentially Settled in, a Company’s Own Stock. Under ASC 480, warrants are considered liability classified if the warrants are mandatorily redeemable, obligate the issuer to settle the warrants or the underlying shares by paying cash or other assets, or warrants that must or may require settlement by issuing variable number of shares. If warrants do not meet liability classification under ASC 480-10, Lineage assesses the requirements under ASC 815-40, which states that contracts that require or may require the issuer to settle the contract for cash are liabilities recorded at fair value, irrespective of the likelihood of the transaction occurring that triggers the net cash settlement feature. If the warrants do not require liability classification under ASC 815-40, in order to conclude equity classification, Lineage assesses whether the warrants are indexed to its common stock or its subsidiary’s common stock, as applicable, and whether the warrants are classified as equity under ASC 815-40 or other applicable GAAP. After all relevant assessments are made, Lineage concludes whether the warrants are classified as liability or equity. Liability classified warrants are required to be accounted for at fair value both on the date of issuance and on subsequent accounting period ending dates, with all changes in fair value after the issuance date recorded in the consolidated statements of operations as a gain or loss. Equity classified warrants are accounted for at fair value on the issuance date with no changes in fair value recognized subsequent to the issuance date. In 2017, Cell Cure issued certain liability classified warrants (see Note 11) and in 2019, Lineage assumed certain warrants in connection with the closing of the Asterias Merger (see Note 3).
| 80 |
Transactions with noncontrolling interests of subsidiaries - Lineage accounts for a change in ownership interests in its subsidiaries that does not result in a change of control of the subsidiary by Lineage under the provisions of ASC 810-10-45-23, Consolidation – Other Presentation Matters, which prescribes the accounting for changes in ownership interest that do not result in a change in control of the subsidiary, as defined by GAAP, before and after the transaction. Under this guidance, changes in a controlling shareholder’s ownership interest that do not result in a change of control, as defined by GAAP, in the subsidiary are accounted for as equity transactions. Thus, if the controlling shareholder retains control, no gain or loss is recognized in the statements of operations of the controlling shareholder. Similarly, the controlling shareholder will not record any additional acquisition adjustments to reflect its subsequent purchases of additional shares in the subsidiary if there is no change of control. Only a proportional and immediate transfer of carrying value between the controlling and the noncontrolling shareholders occurs based on the respective ownership percentages.
Research and development expenses - Research and development expenses consist of costs incurred for company-sponsored, collaborative and contracted research and development activities. These costs include direct and research-related overhead expenses including compensation and related benefits, stock-based compensation, consulting fees, research and laboratory fees, rent of research facilities, amortization of intangible assets, and license fees paid to third parties to acquire patents or licenses to use patents and other technology. Research and development are expensed as incurred. Research and development expenses incurred and reimbursed by grants from third parties approximate the grant income recognized in the consolidated statements of operations.
General and administrative expenses - General and administrative expenses consist of compensation and related benefits, including stock-based compensation, for executive and corporate personnel; professional and consulting fees; and allocated overhead such as facilities and equipment rent and maintenance, insurance costs allocated to general and administrative expenses, costs of patent applications, prosecution and maintenance, stock exchange-related costs, depreciation expense, marketing costs, and other miscellaneous expenses which are allocated to general and administrative expense.
Foreign currency translation adjustments and other comprehensive income or loss - In countries in which Lineage operates where the functional currency is other than the U.S. dollar, assets and liabilities are translated using published exchange rates in effect at the consolidated balance sheet date. Revenues and expenses and cash flows are translated using an approximate weighted average exchange rate for the period. Resulting foreign currency translation adjustments are recorded as other comprehensive income or loss, net of tax, in the consolidated statements of comprehensive income or loss and included as a component of accumulated other comprehensive income or loss on the consolidated balance sheets. Foreign currency translation adjustments are primarily attributable to Cell Cure and ESI, Lineage’s consolidated foreign subsidiaries. For the years ended December 31, 2020 and 2019, comprehensive loss includes foreign currency translation adjustments, net of tax, of $3.0 million and $2.1 million, respectively.
Foreign currency transaction gains and losses - For transactions denominated in other than the functional currency of Lineage or its subsidiaries, Lineage recognizes transaction gains and losses in the consolidated statements of operations and classifies the gain or loss based on the nature of the item that generated it. The majority of Lineage’s foreign currency transaction gains and losses are generated by Cell Cure’s intercompany debt due to Lineage, which are U.S. dollar-denominated, while Cell Cure’s functional currency is the Israeli New Shekel (“ILS”). At each balance sheet date, Lineage remeasures the intercompany debt using the current exchange rate at that date pursuant to ASC 830, Foreign Currency Matters. These foreign currency remeasurement gains and losses are included in other income and expenses, net.
Income taxes - Lineage accounts for income taxes in accordance with ASC 740, Income Taxes, which prescribe the use of the asset and liability method, whereby deferred tax asset or liability account balances are calculated at the balance sheet date using current tax laws and rates in effect. Valuation allowances are established when necessary to reduce deferred tax assets when it is more likely than not that a portion or all of the deferred tax assets will not be realized. ASC 740 guidance also prescribes a recognition threshold and a measurement attribute for the financial statement recognition and measurement of tax positions taken or expected to be taken in a tax return. For benefits to be recognized, a tax position must be more-likely-than-not sustainable upon examination by taxing authorities. Lineage files a U.S. federal income tax return as well as various state and foreign income tax returns. Lineage’s judgments regarding future taxable income may change over time due to changes in market conditions, changes in tax laws, tax planning strategies or other factors. If Lineage assumptions, and consequently the estimates, change in the future with respect to Lineage’s own deferred tax assets and liabilities, the valuation allowance may be increased or decreased, which may have a material impact on Lineage’s consolidated financial statements. Lineage recognizes accrued interest and penalties related to unrecognized tax benefits, if any, as income tax expense; however, no amounts were accrued for the payment of interest and penalties as of December 31, 2020 and 2019.
| 81 |
Although the fair value of employee stock options is determined in accordance with FASB guidance, changes in the assumptions can materially affect the estimated value and therefore the amount of compensation expense recognized in the consolidated financial statements.
Royalties from product sales and license fees - Lineage’s performance obligations in agreements with certain customers is to provide a license to allow customers to make, import and sell company licensed products or methods for preclinical studies and commercial use. Customers pay a combination of a license issue fee paid up front and a sales-based royalty, if any, in some cases with yearly minimums. The transaction price is deemed to be the license issue fee stated in the contract. The license offered by Lineage is a functional license with significant standalone functionality and provides customers with the right to use Lineage’s intellectual property. This allows Lineage to recognize revenue on the license issue fee at a point in time at the beginning of the contract, which is when the customer begins to have use of the license. Variable consideration related to sales-based royalties is recognized only when (or as) the later of one or more of the following events occur: (a) a sale or usage occurs, or (b) the performance obligation to which some, or all, of the sales-based or usage-based royalty that has been allocated and has been satisfied or partially satisfied. Due to the contract termination clauses, Lineage does not expect to receive all of the minimum royalty payments throughout the term of the agreements. Therefore, Lineage fully constrains recognition of the minimum royalty payments as revenues until its customers are obligated to pay, which is generally within 60 days prior to the beginning of each year the minimum royalty payments are due.
Grant revenues - In applying the provisions of Topic 606, Lineage has determined that government grants are out of the scope of Topic 606 because the government entities do not meet the definition of a “customer”, as defined by Topic 606, as there is not considered to be a transfer of control of good or services to the government entities funding the grant. Lineage has, and will continue to, account for grants received to perform research and development services in accordance with ASC 730-20, Research and Development Arrangements, which requires an assessment, at the inception of the grant, of whether the grant is a liability or a contract to perform research and development services for others. If Lineage or a subsidiary receiving the grant is obligated to repay the grant funds to the grantor regardless of the outcome of the research and development activities, then Lineage is required to estimate and recognize that liability. Alternatively, if Lineage or a subsidiary receiving the grant is not required to repay, or if it is required to repay the grant funds only if the research and development activities are successful, then the grant agreement is accounted for as a contract to perform research and development services for others, in which case, grant revenue is recognized when the related research and development expenses are incurred.
Deferred grant revenues represent grant funds received from the governmental funding agencies for which the allowable expenses have not yet been incurred as of the balance sheet date reported.
Revenue Recognition by Source and Geography - Revenues are recognized when control of the promised goods or services is transferred to customers, or in the case of governmental entities funding a grant, when allowable expenses are incurred, in an amount that reflects the consideration Lineage or a subsidiary, depending on which company has the customer or the grant, expects to be entitled to in exchange for those goods or services.
| 82 |
The following table presents Lineage’s consolidated revenues disaggregated by source (in thousands).
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| REVENUES: | ||||||||
| Grant revenue | $ | 1,053 | $ | 2,037 | ||||
| Royalties from product sales and license fees | 773 | 1,221 | ||||||
| Sale of research products and services | 257 | |||||||
| Total revenues | $ | 1,826 | $ | 3,515 | ||||
The following table presents consolidated revenues, disaggregated by geography, based on the billing addresses of customers, or in the case of grant revenues, based on where the governmental entities that fund the grant are located (in thousands).
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| REVENUES: | ||||||||
| United States | $ | 1,160 | $ | 2,092 | ||||
| Foreign (1) | 666 | 1,423 | ||||||
| Total revenues | $ | 1,826 | $ | 3,515 | ||||
| (1) |
Recently Adopted Accounting Pronouncements
In August 2018, the FASB issued ASU 2018-13, Fair Value Measurement (Topic 820): Disclosure Framework – Changes to the Disclosure Requirements for Fair Value Measurement, which modifies certain disclosure requirements for reporting fair value measurements. ASU 2018-13 is effective for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2019. Lineage adopted this standard on January 1, 2020 and it did not have a significant impact on our consolidated financial statements.
Recently Issued Accounting Pronouncements Not Yet Adopted - The following accounting standards, which are not yet effective, are presently being evaluated by Lineage to determine the impact that they might have on its consolidated financial statements.
In December 2019, the FASB issued ASU 2019-12, Simplifying the Accounting for Income Taxes. The ASU enhances and simplifies various aspects of the income tax accounting guidance in ASC 740 and removes certain exceptions for recognizing deferred taxes for investments, performing intraperiod allocation and calculating income taxes in interim periods. The ASU also adds guidance to reduce complexity in certain areas, including recognizing deferred taxes for tax goodwill and allocating taxes to members of a consolidated group. This ASU is effective for fiscal years beginning after December 15, 2020, and interim periods within those fiscal years with early adoption permitted. Lineage adopted this standard as of January 1, 2021 and it is not expected to have a material impact on the consolidated financial statements.
In June 2016, the FASB issued ASU 2016-13, Financial Instruments – Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments. ASU 2016-13 is intended to provide financial statement users with more decision-useful information about the expected credit losses on financial instruments and other commitments and requires consideration of a broader range of reasonable and supportable information to inform credit loss estimates. ASU 2016-13 is effective for Lineage beginning January 1, 2023. Lineage has not yet completed its assessment of the impact of the new standard on its consolidated financial statements.
3. Asterias Merger
On March 8, 2019, the Asterias Merger closed with Asterias surviving as a wholly owned subsidiary of Lineage. The former stockholders of Asterias (other than Lineage) received common shares of Lineage (the “Merger Consideration”) for every share of Asterias common stock they owned (the “Merger Exchange Ratio”). Lineage issued common shares, including shares issued in respect of restricted stock units issued by Asterias that immediately vested in connection with the closing of the Asterias Merger. The fair value of such shares, based on the closing price of Lineage common shares on March 8, 2019, was $32.4 million.
| 83 |
In connection with the closing of the Asterias Merger, Lineage assumed outstanding warrants to purchase shares of Asterias common stock, as further discussed below and in Note 11, and assumed sponsorship of the Asterias 2013 Equity Incentive Plan (see Note 12). All stock options to purchase shares of Asterias common stock outstanding immediately prior to the closing of the Asterias Merger were canceled at the closing for no consideration.
As of December 31, 2019, the assets and liabilities of Asterias have been included in the consolidated balance sheet of Lineage. The results of operations of Asterias from March 8, 2019 through December 31, 2019 have been included in the consolidated statement of operations of Lineage for the year ended December 31, 2019.
Calculation of the purchase price
The calculation of the purchase price for the Asterias Merger and the Merger Consideration transferred on March 8, 2019 was as follows (in thousands, except for share and per share amounts):
Lineage (38%
interest) | Shareholders other than Lineage (approximate 62% ownership interest) | Total | ||||||||||
| Outstanding Asterias common stock as of March 8, 2019 | 21,747,569 | 34,783,333 | (1) | 56,530,902 | (1) | |||||||
| Exchange ratio | 0.710 | 0.710 | 0.710 | |||||||||
| Lineage common shares issuable | 15,440,774 | (2) | 24,695,898 | (3) | 40,136,672 | |||||||
| Per share price of Lineage common shares as of March 8, 2019 | $ | 1.31 | $ | 1.31 | $ | 1.31 | ||||||
| Purchase price (in $000s) | $ | 20,227 | (2) | $ | 32,353 | $ | 52,580 | |||||
| (1) | |
| (2) | |
| (3) |
Purchase price allocation
Lineage allocated the acquisition consideration to tangible and identifiable intangible assets acquired and liabilities assumed based on their estimated fair values as of the acquisition date. The fair value of the acquired tangible and identifiable intangible assets were determined based on inputs that are unobservable and significant to the overall fair value measurement. It is also based on estimates and assumptions made by management at the time of the acquisition. As such, this was classified as Level 3 fair value hierarchy measurements and disclosures.
| 84 |
The allocation of the purchase price in the table below is based on our estimates of the fair values of tangible and intangible assets acquired, including IPR&D, and liabilities assumed as of the acquisition date, with the excess recorded as goodwill (in thousands). As of December 31, 2019, Lineage had finalized its purchase price allocation.
| Assets acquired: | ||||
| Cash and cash equivalents | $ | 3,117 | ||
| Prepaid expenses and other assets, current and noncurrent | 660 | |||
| Machinery and equipment | 308 | |||
| Long-lived intangible assets - royalty contracts | 650 | |||
| Acquired in-process research and development (“IPR&D”) | 46,540 | |||
| Total assets acquired | 51,275 |
| Liabilities assumed: | ||||
| Accrued liabilities and accounts payable | 982 | |||
| Liability classified warrants | 867 | |||
| Deferred license revenue | 200 | |||
| Long-term deferred income tax liability | 10,753 | |||
| Total liabilities assumed | 12,802 | |||
| Net assets acquired, excluding goodwill (a) | 38,473 | |||
| Fair value of Lineage common shares held by Asterias (b) | 3,435 | |||
| Total purchase price (c) | 52,580 | |||
| Estimated goodwill (c-a-b) | $ | 10,672 |
The valuation of identifiable intangible assets and their estimated useful lives are as follows (in thousands, except for useful life):
| Preliminary Estimated Asset Fair Value |
Useful Life (Years) | |||||||
| (in thousands, except for useful life) | ||||||||
| In process research and development (“IPR&D”) | $ | 46,540 | n/a | |||||
| Royalty contracts | 650 | 5 | ||||||
| $ | 47,190 | |||||||
The following is a discussion of the valuation methods used to determine the fair value of Asterias’ significant assets and liabilities in connection with the Asterias Merger:
IPR&D and Deferred Income Tax Liability - The fair value of identifiable acquired IPR&D intangible assets consisting of $31.7 million pertaining to the OPC1 program that is currently in a Phase 1/2a clinical trial for SCI, which has been partially funded by the California Institute for Regenerative Medicine and $14.8 million pertaining to the VAC2 program, which is an allogeneic, or “off-the-shelf,” cancer immunotherapy derived from pluripotent stem cells for which a clinical trial in non-small cell lung cancer is being funded and sponsored by Cancer Research UK. The identification of these intangible assets are based on consideration of historical experience and a market participant’s view further discussed below; collectively, OPC1 and VAC2 are referred to as the “AST-Clinical Programs”. These intangible assets are valued primarily through the use of a probability weighted discounted cash flow method under the income approach further discussed below. Lineage considered Asterias’ VAC1 program, which is an autologous, or patient-specific, cancer immunotherapy derived from the patient’s own cells, to have de minimis value due to significant risks, substantial costs and limited opportunities.
| 85 |
Lineage determined that the estimated aggregate fair value of the AST-Clinical programs was $46.5 million as of the acquisition date using a probability weighted discounted cash flow method for each respective program. This approach estimates the probability of the AST-Clinical Programs achieving successful completion of remaining clinical trials and related approvals into the valuation technique.
To calculate fair value of the AST-Clinical programs under the discounted cash flow method, Lineage used probability-weighted, projected cash flows discounted at a rate considered appropriate given the significant inherent risks associated with cell therapy development by clinical-stage companies. Cash flows were calculated based on estimated projections of revenues and expenses related to each respective program. Cash flows were assumed to extend through a seven-year market exclusivity period for the OPC1 program from the date of market launch. Revenues from commercialization of the AST-Clinical Programs were based on estimated market potential for the indication of each program. The resultant cash flows were then discounted to present value using a weighted-average cost of capital for companies with profiles substantially similar to that of Lineage, which Lineage believes represents the rate that market participants would use to value the assets. Lineage compensated for the phase of development of the program by applying a probability factor to its estimation of the expected future cash flows. The projected cash flows were based on significant assumptions, including the indications in which Lineage will pursue development of the AST-Clinical programs, the time and resources needed to complete the development and regulatory approval, estimates of revenue and operating profit related to the program considering its stage of development, the life of the potential commercialized product, market penetration and competition, and risks associated with achieving commercialization, including delay or failure to obtain regulatory approvals to conduct clinical studies, failure of clinical studies, delay or failure to obtain required market clearances, and intellectual property litigation.
These IPR&D assets are indefinite-lived intangible assets until the completion or abandonment of the associated research and development (“R&D”) efforts. Once the R&D efforts are completed or abandoned, the IPR&D will either be amortized over the asset life as a finite-lived intangible asset or be impaired, respectively, in accordance with ASC 350, Intangibles - Goodwill and Other. In accordance with ASC 350, goodwill and acquired IPR&D are determined to have indefinite lives and, therefore, are not amortized. Instead, they are tested for impairment at least annually and between annual tests if Lineage becomes aware of an event or a change in circumstances that would indicate the asset may be impaired.
Because the IPR&D (prior to completion or abandonment of the R&D) is considered an indefinite-lived asset for accounting purposes, the fair value of the IPR&D on the acquisition date creates a deferred income tax liability (“DTL”) in accordance with ASC 740, Income Taxes (see Note 13). This DTL is computed using the fair value of the IPR&D assets on the acquisition date multiplied by Lineage’s federal and state income tax rates. While this DTL would reverse on impairment or sale or commencement of amortization of the related intangible assets, those events are not anticipated under ASC 740 for purposes of predicting reversal of a temporary difference to support the realization of deferred tax assets, except for certain deferred tax assets and credit carryforwards that are also indefinite in nature as of the closing of the Asterias Merger, which may be considered for reversal under ASC 740 as further discussed in Note 13.
Royalty contracts – Asterias has certain royalty revenues for “research only use” culture media for preclinical research applications under certain, specific patent families under contracts which preclude the customers to sell for commercial use or for clinical trials. These royalty cash flows are generated under certain specific patent families which Asterias previously acquired from Geron Corporation (“Geron”). Asterias pays Geron a royalty for all royalty revenues received from these contracts. Because these patents are a subset of the clinical programs discussed above, are expected to continue to generate revenues for Asterias and are not to be used in the OPC1 or the VAC2 programs, these patents are considered to be separate long-lived intangible assets under ASC 805. These intangible assets are also valued primarily through the use of the discounted cash flow method under the income approach, and will be amortized over their useful life, estimated to be 5 years. The discounted cash flow method estimated the amount of net royalty income that can be expected under the contracts in future years. The amounts were based on observed historical trends in the growth of these revenue streams, and were estimated to terminate in approximately five years, when the key patents under these contracts will begin to expire. The resulting cash flows were discounted to the valuation date based on a rate of return that recognizes a lower level of risk associated with these assets as compared to the AST-Clinical programs discussed above.
| 86 |
Deferred license revenue – In September 2018, Asterias and Novo Nordisk A/S (“Novo Nordisk”) entered into an option for Novo Nordisk or its designated U.S. affiliate to license, on a non-exclusive basis, certain intellectual property related to culturing pluripotent stem cells, such as hES cells, in suspension. Under the terms of the option, Asterias received a one-time upfront payment of $1.0 million, in exchange for a 24-month period option to negotiate a non-exclusive license during which time Asterias has agreed to not grant any exclusive licenses inconsistent with the Novo Nordisk option. This option is considered a performance obligation as it provides Novo Nordisk with a material right that it would not receive without entering into the contract.
For business combination purposes under ASC 805, the fair value of this performance obligation to Lineage, from a market participant perspective, is the estimated costs Lineage may incur, plus a normal profit margin for the level of effort required to perform under the contract after the acquisition date, assuming Novo Nordisk exercised its option, including, but not limited to, negotiation costs, legal fees, arbitration, if any, and other related costs. Management has estimated those costs, plus a normal profit margin, to be approximately $200,000 in the purchase price allocation. This amount was originally recorded as deferred revenue and subsequently recognized as revenue in September 2020 when Novo Nordisk did not exercise the option.
Liability classified warrants – On May 13, 2016, in connection with a common stock offering, Asterias issued warrants to purchase 2,959,559 shares of Asterias common stock (the “Asterias Warrants”) with an exercise price of $4.37 per share that expire in years from the issuance date, or May 13, 2021. As of the closing of the Asterias Merger, there were 2,813,159 Asterias Warrants outstanding. The Asterias Warrants contain certain provisions in the event of a Fundamental Transaction, as defined in the warrant agreement governing the Asterias Warrants (“Warrant Agreement”), that Asterias or any successor entity will be required to purchase, at a holder’s option, exercisable at any time concurrently with or within thirty days after the consummation of the fundamental transaction, the Asterias Warrants for cash in an amount equal to the calculated value of the unexercised portion of such holder’s warrants, determined in accordance with the Black-Scholes option pricing model with significant inputs as specified in the Warrant Agreement. The Asterias Merger was a Fundamental Transaction for purposes of the Asterias Warrants.
The fair value of the Asterias Warrants was determined by using Black-Scholes option pricing models which take into consideration the probability of the Fundamental Transaction, which for purposes of the above valuation was assumed to be at 100% and net cash settlement occurring, using the contractual remaining term of the warrants. In applying these models, these inputs included key assumptions including the per share closing price of Lineage common shares on March 8, 2019, volatility computed in accordance with the provisions of the Warrant Agreement and, to a large extent, assumptions based on discussions with a majority of the holders of the Asterias Warrants since the closing of the Asterias Merger to settle the Asterias Warrants in cash or in common shares of Lineage. Based on such discussions, Lineage believes the fair value of the Asterias Warrants as of the closing of the Asterias Merger is not subject to change significantly, however, to the extent any Asterias Warrants that were not settled in cash or in Lineage common shares discussed below, were automatically converted to Lineage warrants 30 days after the closing of the Asterias Merger. In April 2019, Asterias Warrants representing approximately $372,000 in fair value were settled: $332,000 in fair value was settled in exchange for common shares of Lineage, and $40,000 in fair value was settled in exchange for cash. The Asterias Warrants settled in exchange for common shares of Lineage were held by Broadwood Partners, L.P., an Asterias and Lineage shareholder. The Asterias Warrants settled in exchange for cash were held by other parties. The remaining Asterias Warrants (representing approximately $495,000 in fair value as of March 31, 2019) were converted into warrants to purchase common shares of Lineage using the Merger Exchange Ratio (the “Lineage Warrants”).
As of December 31, 2020, the total number of common shares of Lineage subject to warrants that were assumed by Lineage in connection with the Asterias Merger was 1,089,900, with similar terms and conditions retained under the Lineage Warrants as per the original Warrant Agreements. The Lineage Warrants have an exercise price of $6.15 per warrant share and expire on May 13, 2021. Lineage is accounting for the outstanding Lineage Warrants as a liability at fair value, with subsequent changes to the fair value of the Lineage Warrants at each reporting period thereafter included in the consolidated statement of operations (see Note 11).
| 87 |
Fair value of Lineage common shares held by Asterias – As of March 8, 2019, Asterias held 2,621,811 common shares of Lineage as marketable securities on its standalone financial statements. The fair value of those shares acquired by Lineage from Asterias is determined based on the $ per share closing price of Lineage common shares on March 8, 2019. Although treasury shares are not considered an asset and were retired upon Lineage’s acquisition of Asterias, the fair value of those shares is a part of the purchase price allocation shown in the tables above. These Lineage shares were retired at the completion of the Asterias Merger.
Goodwill – Goodwill is calculated as the difference between the acquisition date fair value of the consideration transferred and the values assigned to the assets acquired and liabilities assumed. Goodwill is not amortized but is tested for impairment at least annually, or more frequently if circumstances indicate potential impairment.
Depending on the structure of a particular acquisition, goodwill and identifiable intangible assets may not be deductible for tax purposes. Goodwill recorded in the Asterias Merger is not expected to be deductible for tax purposes (see Note 13).
During the years ended December 31, 2020 and 2019, Lineage incurred $0.7 million and $5.1 million, respectively, in acquisition related costs which were recorded in general and administrative expenses in the accompanying consolidated statements of operations.
Prior to the Asterias Merger being consummated in March 2019, Lineage elected to account for its million shares of Asterias common stock at fair value using the equity method of accounting. The fair value of the Asterias shares was approximately $20.2 million as of March 8, 2019, the closing date of the Asterias Merger, based on $0.93 per share, which was calculated by multiplying (a) $1.31, the closing price of Lineage common shares on such date by (b) the Merger Exchange Ratio. The fair value of the Asterias shares was approximately $13.5 million as of December 31, 2018, based on the closing price of Asterias common stock of $ per share on such date. Accordingly, Lineage recorded an unrealized gain of $6.7 million for the year ended December 31, 2019, representing the change in fair value of Asterias common stock from December 31, 2018 to March 8, 2019. All share prices were determined based on the closing price of Lineage or Asterias common stock on the NYSE American on the applicable dates.
Asterias Merger Related Litigation – See Note 14 Commitments and Contingencies for discussion regarding litigation related to the Asterias Merger.
4. Accounting for Common Stock of OncoCyte, at Fair Value
Prior to September 11, 2019, Lineage elected to account for its shares of OncoCyte common stock at fair value using the equity method of accounting. Lineage sold million shares of OncoCyte common stock for net proceeds of $4.2 million in July 2019. Accordingly, Lineage’s ownership in OncoCyte was reduced from 28% to 24%. Lineage sold an additional million shares of OncoCyte common stock for net proceeds of $6.5 million on September 11, 2019. Lineage’s ownership in OncoCyte was further reduced to 16% at this time. Effective September 11, 2019, Lineage began accounting for its shares of OncoCyte common stock as marketable equity securities. The calculation of fair value is the same under the equity method and as a marketable equity security.
As of December 31, 2019, we had million shares of OncoCyte common stock. These shares had a fair value of $19.0 million, based on the closing price of OncoCyte common stock of $ per share on December 31, 2019.
During the year ended December 31, 2020, Lineage sold approximately million shares of OncoCyte common stock for net proceeds of $10.9 million.
As of December 31, 2020, we owned million shares of OncoCyte common stock. These shares had a fair value of $8.7 million, based on the closing price of OncoCyte common stock of $ per share on December 31, 2020.
For the year ended December 31, 2020, we recorded a realized gain of $3.1 million due to sales of OncoCyte shares in the period. In the same period, we also recorded an unrealized loss of $2.5 million related to its OncoCyte shares. The unrealized loss is comprised of $3.7 million related to the difference between the book cost basis of OncoCyte shares sold in the period versus the applicable prior month’s ending OncoCyte stock price, which is offset by $ million related to the shares remaining at December 31, 2020 and the increase in OncoCyte’s stock price from $2.25 at December 31, 2019 to $ at December 31, 2020.
| 88 |
For the year ended December 31, 2019, we recorded a realized gain of $0.5 million due to sales of OncoCyte shares in the period. We also recorded an unrealized gain of $8.8 million due to the increase in OncoCyte’s stock price from $ per share at December 31, 2018 to $2.25 per share at December 31, 2019. $8.0 million of the unrealized gain was recorded as an unrealized gain on an equity method investment as it was prior to September 11, 2019; the remaining $0.8 million was recorded as an unrealized gain on marketable equity securities.
All share prices are determined based on the closing price of OncoCyte common stock on the NYSE American on the applicable dates, or the last day of trading of the applicable quarter, if the last day of a quarter fell on a weekend.
5. Sale of Significant Ownership Interest in AgeX to Juvenescence Limited
On August 30, 2018, Lineage entered into a Stock Purchase Agreement with Juvenescence Limited and AgeX, pursuant to which Lineage sold million shares of common stock of AgeX to Juvenescence for $ per share, or an aggregate purchase price of $43.2 million (the “Purchase Price”). Juvenescence paid $10.8 million of the Purchase Price at closing, issued an unsecured convertible promissory note dated August 30, 2018 in favor of Lineage for $21.6 million (the “Promissory Note”), and paid $10.8 million on November 2, 2018. The Stock Purchase Agreement contains customary representations, warranties and indemnities from Lineage relating to the business of AgeX, including an indemnity cap of $4.3 million, which is subject to certain exceptions. In connection with the sale, Lineage also entered into a Shared Facilities Agreement with AgeX (see Note 10).
The Promissory Note bore interest at 7% per annum, with principal and accrued interest payable at maturity on August 30, 2020. The Promissory Note was paid in full for a total of $24.6 million on August 28, 2020.
For the years ended December 31, 2020, and 2019, Lineage recognized $1,008,000 and $1,512,000, respectively, in interest income on the Promissory Note.
The Shared Facilities Agreement was terminated on July 31, 2019 with respect to the use of Lineage’s office and laboratory facilities and September 30, 2019 with respect to all other remaining shared services.
6. Property and Equipment, Net
At December 31, 2020 and 2019, property and equipment, net were comprised of the following (in thousands):
| December 31, | ||||||||
| 2020 | 2019 | |||||||
| Equipment, furniture and fixtures | $ | 3,628 | $ | 4,148 | ||||
| Leasehold improvements | 2,472 | 2,862 | ||||||
| Right-of-use assets (1) | 3,845 | 5,756 | ||||||
| Accumulated depreciation and amortization | (4,315 | ) | (4,591 | ) | ||||
| Property and equipment, net | $ | 5,630 | $ | 8,175 | ||||
| (1) |
Property and equipment at December 31, 2020 and 2019 includes $79,000 and $96,000 financed by capital leases, respectively. In September 2020, Lineage terminated its leases in Alameda and entered into a new lease for a reduced amount of square footage. This resulted in a net reduction to right-of-use assets of approximately $1.4 million. In December 2020, Cell Cure extended ones of its leases (“the Original Cell Cure Lease”) for an additional five years, which resulted in a net increase to right-of-use assets of $0.6 million. See additional information in Note 14.
Depreciation and amortization expense amounted to $0.9 million and $1.1 million for the years ended December 31, 2020 and 2019, respectively.
| 89 |
During the year ended December 31, 2020, Lineage sold equipment with a net book value of $32,000 and recognized a loss of $9,000. Lineage also wrote off assets with net book values of $156,000, with $104,000 of this amount related to the termination of its leases in Alameda. Additionally, Lineage sold non-capitalized assets for a net gain of $72,000.
During the year ended December 31, 2019, Lineage sold equipment with a net book value of $209,000 and recognized a loss of $109,000. Primarily in connection with the close out of the Asterias facility, Lineage also sold non-capitalized assets for a net gain of $337,000.
Gains related to the sale of assets are included in research and development expenses on the statement of operations. Write offs of assets are included in other income, net on the statement of operations.
7. Goodwill and Intangible Assets, Net
At December 31, 2020 and 2019, goodwill and intangible assets, net consisted of the following: (in thousands):
| December 31, | ||||||||
| 2020 | 2019 | |||||||
| Goodwill (1) | $ | 10,672 | $ | 10,672 | ||||
| Intangible assets: | ||||||||
| Acquired IPR&D – OPC1 (from the Asterias Merger) (2) | $ | 31,700 | $ | 31,700 | ||||
| Acquired IPR&D – VAC2 (from the Asterias Merger) (2) | 14,840 | 14,840 | ||||||
| Intangible assets subject to amortization: | ||||||||
| Acquired patents | 18,953 | 18,953 | ||||||
| Acquired royalty contracts (2) | 650 | 650 | ||||||
| Total intangible assets | 66,143 | 66,143 | ||||||
| Accumulated amortization | (19,111 | ) | (17,895 | ) | ||||
| Intangible assets, net | $ | 47,032 | $ | 48,248 | ||||
| (1) | |
| (2) |
Lineage amortizes its intangible assets over an estimated period of 5 to 10 years on a straight-line basis. Lineage recognized $1.2 million and $2.0 million in amortization expense of intangible assets during the years ended December 31, 2020 and 2019, respectively.
Amortization of intangible assets for periods subsequent to December 31, 2020 is as follows (in thousands):
| Year Ended December 31, | Amortization Expense | |||
| 2021 | $ | 210 | ||
| 2022 | 130 | |||
| 2023 | 130 | |||
| 2024 | 22 | |||
| Total | $ | 492 | ||
| 90 |
8. Accounts Payable and Accrued Liabilities
At December 31, 2020 and 2019, accounts payable and accrued liabilities consist of the following (in thousands):
| December 31, | ||||||||
| 2020 | 2019 | |||||||
| Accounts payable | $ | 2,611 | $ | 2,427 | ||||
| Accrued compensation | 1,959 | 1,549 | ||||||
| Accrued liabilities | 1,711 | 1,246 | ||||||
| PPP loan payable | 523 | - | ||||||
| Other current liabilities | 9 | 4 | ||||||
| Total | $ | 6,813 | $ | 5,226 | ||||
Accrued liabilities includes $1.0 million related to the signature fee owed to Cancer Research UK, as described in Note 14.
PPP Loan Payable
In April 2020, Lineage received a loan for $523,305 from Axos Bank under the PPP contained within the new Coronavirus Aid, Relief and Economic Security (“CARES”) Act. The PPP loan has a term of two years, is unsecured, and is guaranteed by the U.S. Small Business Administration (“SBA”). The loan carries a fixed interest rate of one percent per annum, with the first six months of interest deferred. Under the CARES Act and Paycheck Protection Program Flexibility Act, Lineage will be eligible to apply for forgiveness of all loan proceeds used to pay payroll costs, rent, utilities and other qualifying expenses during the 24-week period following receipt of the loan, provided that Lineage maintains its employment and compensation within certain parameters during such period. Not more than 40% of the forgiven amount may be for non-payroll costs. If the conditions outlined in the PPP loan program are adhered to by Lineage, all or part of such loan could be forgiven. Lineage believes that all or a substantial portion of the PPP loan is eligible for forgiveness within one year and classifies the loan as a short-term liability. On December 27, 2020, the Consolidated Appropriations Act, 2021 (CAA) was signed into law, retroactively allowing a deduction of the expenses that gave rise to the PPP loan forgiveness, that was previously denied under the CARES Act. California has partially adopted the federal tax treatment. On February 17, 2021, California issued an Immediate Action Agreement, allowing companies to deduct up to $150,000 in expenses covered by the PPP loan. However, Lineage cannot provide any assurance whether the PPP loan will ultimately be forgiven by the SBA. Any forgiven amounts will not be included in Lineage’s taxable income for federal or California purposes. Lineage applied for full forgiveness of the PPP loan on September 30, 2020.
2019 Separation Payments
In connection with the Asterias Merger, several Asterias employees were terminated as of the Asterias Merger date. Three of these employees had employment agreements with Asterias which entitled them to change in control and separation payments in the aggregate of $2.0 million, which such conditions were met on the Asterias Merger date. Accordingly, $2.0 million was accrued and recorded in general and administrative expenses on the merger date and paid in April 2019.
Additionally, Lineage entered into a plan of termination with substantially all other previous employees of Asterias with potential separation payments in the aggregate of $0.5 million. Termination dates for these individuals ranged from May 31, 2019 to June 28, 2019. These employees were required to provide services related to the transition and be an employee of the combined company as of their date of termination in order to receive separation benefits. Since the employees were required to render future services after the merger date, Lineage recorded the aggregate liability ratably over their respective service periods from the Asterias Merger date through the above termination dates, in accordance with ASC 420, Exit or Disposal Cost Obligations. All payments were completed by July 31, 2019.
In connection with the relocation of Lineage’s corporate headquarters to Carlsbad, California, Lineage entered into a plan of termination with certain Lineage employees with potential separation payments in the aggregate of $0.7 million. Termination dates for these individuals range from August 9, 2019 to September 30, 2019. These employees had to provide services related to the transition of services and activities in connection with the relocation and be an employee of Lineage as of their date of termination in order to receive separation benefits. Lineage recorded the aggregate liability ratably over their respective service periods from June 2019 through the above termination dates, in accordance with ASC 420. As of December 31, 2019, all separation payments had been made.
| 91 |
9. Fair Value Measurements
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. To increase the comparability of fair value measures, the following hierarchy prioritizes the inputs to valuation methodologies used to measure fair value (ASC 820-10-50), Fair Value Measurements and Disclosures:
| ● | Level 1 – Inputs to the valuation methodology are quoted prices for identical assets or liabilities in active markets. | |
| ● | Level 2 – Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. | |
| ● | Level 3 – Inputs to the valuation methodology are unobservable; that reflect management’s own assumptions about the assumptions market participants would make and significant to the fair value. |
We measure cash, cash equivalents, marketable securities and our liability classified warrants at fair value on a recurring basis. The fair values of such assets were as follows for December 31, 2020 and 2019 (in thousands):
| Fair Value Measurements Using | ||||||||||||||||
| Balance at December 31, 2020 | Quoted Prices in Active Markets for Identical Assets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | |||||||||||||
| Assets: | ||||||||||||||||
| Cash and cash equivalents | $ | 32,585 | $ | 32,585 | $ | - | $ | |||||||||
| Marketable securities | 8,977 | 8,977 | ||||||||||||||
| Liabilities: | ||||||||||||||||
| Lineage Warrants | 1 | 1 | ||||||||||||||
| Cell Cure Warrants | 437 | 437 | ||||||||||||||
| Fair Value Measurements Using | ||||||||||||||||
| Balance at December 31, 2019 | Quoted Prices in Active Markets for Identical Assets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | |||||||||||||
| Assets: | ||||||||||||||||
| Cash and cash equivalents | $ | 9,497 | $ | 9,497 | $ | - | $ | |||||||||
| Marketable securities | 21,219 | 21,219 | ||||||||||||||
| Liabilities: | ||||||||||||||||
| Lineage Warrants | 20 | 20 | ||||||||||||||
| Cell Cure Warrants | 257 | 257 | ||||||||||||||
| 92 |
We have not transferred any instruments between the three levels of the fair value hierarchy.
In determining fair value, Lineage utilizes valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible, and also considers counterparty credit risk in its assessment of fair value.
Marketable securities include our positions in OncoCyte and HBL. These securities have readily determinable fair values quoted on the NYSE American or TASE stock exchanges. These securities are measured at fair value and reported as current assets on the consolidated balance sheets based on the closing trading price of the security as of the date being presented.
The fair value of Lineage’s assets and liabilities, which qualify as financial instruments under FASB guidance regarding disclosures about fair value of financial instruments, approximate the carrying amounts presented in the accompanying consolidated balance sheets. The carrying amounts of accounts receivable, prepaid expenses and other current assets, accounts payable, accrued expenses and other current liabilities approximate fair values because of the short-term nature of these items.
10. Related Party Transactions
Shared Facilities and Service Agreements with Affiliates
Under the terms of the Shared Facilities Agreements, Lineage allowed OncoCyte and AgeX to use Lineage’s premises and equipment located at Lineage’s headquarters in Alameda, California for the purpose of conducting business. Lineage also provided accounting, billing, bookkeeping, payroll, treasury, payment of accounts payable, and other similar administrative services to OncoCyte and AgeX. The Shared Facilities Agreements also allowed Lineage to provide the services of attorneys, accountants, and other professionals who may provide professional services to Lineage. Lineage also provided OncoCyte and AgeX with the services of laboratory and research personnel, including Lineage employees and contractors, for the performance of research and development work for OncoCyte and AgeX at the premises. Shared services with AgeX were terminated on July 31, 2019 with respect to the use of Lineage’s office and laboratory facilities and September 30, 2019 with respect to all other remaining shared services. Shared services with OncoCyte were terminated on September 30, 2019, and December 31, 2019 with respect to all other remaining shared services.
Lineage charged OncoCyte and AgeX a “Use Fee” for services provided and for use of Lineage facilities, equipment, and supplies. For each billing period, Lineage prorated and allocated to OncoCyte and AgeX costs incurred, including costs for services of Lineage employees and use of equipment, insurance, leased space, professional services, software licenses, supplies and utilities. The allocation of costs depended on key cost drivers, including actual documented use, square footage of facilities used, time spent, costs incurred by Lineage for OncoCyte and AgeX, or upon proportionate usage by Lineage, OncoCyte and AgeX, as reasonably estimated by Lineage. Lineage, at its discretion, had the right to charge OncoCyte and AgeX a 5% markup on such allocated costs. The allocated cost of Lineage employees and contractors who provided services was based upon the number of hours or estimated percentage of efforts of such personnel devoted to the performance of services.
The Use Fee was determined and invoiced to OncoCyte and AgeX on a regular basis, generally monthly or quarterly. Each invoice was payable in full within 30 days after receipt. Any invoice, or portion thereof, not paid in full when due bore interest at the rate of 15% per annum until paid, unless the failure to make a payment was due to any inaction or delay in making a payment by Lineage. Lineage did not charge OncoCyte or AgeX any interest.
In addition to the Use Fee, OncoCyte and AgeX reimbursed Lineage for any out of pocket costs incurred by Lineage for the purchase of office supplies, laboratory supplies, and other goods and materials and services for the account or use of OncoCyte or AgeX. Lineage was not obligated to purchase or acquire any office supplies or other goods and materials or any services for OncoCyte or AgeX, and if any such supplies, goods, materials or services were obtained, Lineage could arrange for the suppliers to invoice OncoCyte or AgeX directly.
| 93 |
The Use Fees charged to OncoCyte and AgeX shown above were not reflected in revenues, but instead Lineage’s general and administrative expenses and research and development expenses are shown net of those charges in the consolidated statements of operations.
For the year ended December 31, 2019, Lineage charged Use Fees of $2,176,000 to OncoCyte and AgeX; $890,000 was offset against general and administrative expenses and $1,286,000 was offset against research and development expenses.
Other related party transactions
Lineage currently pays $5,050 per month for the use of approximately 900 square feet of office space in New York City, which is made available to Lineage on a month-by-month basis by one of its directors at an amount that approximates his cost (see Note 14). These payments are expected to cease in March 2021 when the office space lease expires.
In April 2019, Lineage issued common shares of Lineage to Broadwood Partners, L.P., an Asterias and Lineage shareholder, in exchange for the settlement of Asterias Warrants in connection with the Asterias Merger (see Note 3).
In connection with the putative shareholder class action lawsuits filed in February 2019 and October 2019 challenging the Asterias Merger (see Note 14), Lineage has agreed to pay for the legal defense of Neal Bradsher, director, and Broadwood Partners, L.P., a shareholder of Lineage, and Broadwood Capital, Inc., which manages Broadwood Partners, L.P., all of which were named in the lawsuits. Through December 31, 2020, Lineage has incurred a total of $359,000 in legal expenses on behalf of the director, shareholder and the manager of the shareholder.
As part of financing transactions in which there were multiple other purchasers, Broadwood Partners, L.P. purchased shares, shares and shares of OncoCyte common stock from Lineage in July 2019, September 2019 and January 2020, respectively.
11. Shareholders’ Equity
Preferred Shares
Lineage is authorized to issue shares of preferred stock. The preferred shares may be issued in one or more series as the board of directors may by resolution determine. The board of directors is authorized to fix the number of shares of any series of preferred shares and to determine or alter the rights, preferences, privileges, and restrictions granted to or imposed on the preferred shares as a class, or upon any wholly unissued series of any preferred shares. The board of directors may, by resolution, increase or decrease (but not below the number of shares of such series then outstanding) the number of shares of any series of preferred shares subsequent to the issue of shares of that series. As of December 31, 2020, shares of preferred stock were issued or outstanding.
Common Shares
At December 31, 2020, Lineage was authorized to issue common shares, no par value. As of December 31, 2020 and 2019, Lineage had and issued and outstanding common shares, respectively.
During the years ended December 31, 2020 and 2019, Lineage issued and common shares, net of shares withheld and retired for employee taxes paid, respectively, for vested restricted stock units (see Note 12).
| 94 |
At-the-Market (“ATM”) Offering
On May 1, 2020, Lineage entered into the Sales Agreement, pursuant to which Lineage may offer and sell, from time to time, through Cantor Fitzgerald, common shares of Lineage (“ATM Shares”) having an aggregate offering price of up to $25,000,000. Lineage is not obligated to sell any ATM Shares. Subject to the terms and conditions of the Sales Agreement, Cantor Fitzgerald will use commercially reasonable efforts, consistent with its normal trading and sales practices, applicable state and federal law, rules and regulations, and the rules of the NYSE American, to sell the ATM Shares from time to time based upon Lineage’s instructions, including any price, time or size limits specified by Lineage. Under the Sales Agreement, Cantor Fitzgerald may sell the ATM Shares by any method deemed to be an “at-the-market” offering as defined in Rule 415(a)(4) under the Securities Act of 1933, as amended, or by any other method permitted by law, including in privately negotiated transactions. Cantor Fitzgerald’s obligations to sell the ATM Shares are subject to satisfaction of certain conditions, including the continued effectiveness of Lineage’s Registration Statement on Form S-3 (File No. 333-237975), which was filed with the Commission on May 1, 2020 and was declared effective on May 8, 2020. The Sales Agreement replaced the previous sales agreement with Cantor that had been entered into in April 2017. As of December 31, 2020, Lineage sold ATM Shares for gross and net proceeds of $5.1 million and $5.0 million, respectively (in each case, which excludes $0.3 million of cash in transit related to 2020 sales that settled in 2021). In the first quarter of 2021 through March 5, 2021, Lineage sold an additional ATM Shares for gross and net proceeds of $million and $million, respectively (in each case, which includes $ million of cash in transit related to 2020 sales that settled in 2021). On March 5, 2021, Lineage filed a prospectus supplement with the SEC in connection with the offer and sale of an additional $million of ATM Shares under the Sales Agreement.
Lineage agreed to pay Cantor Fitzgerald a commission of 3.0% of the aggregate gross proceeds from each sale of shares, reimburse legal fees and disbursements and provide Cantor Fitzgerald with customary indemnification and contribution rights. The Sales Agreement may be terminated by Cantor Fitzgerald or Lineage at any time upon notice to the other party, or by Cantor Fitzgerald at any time in certain circumstances, including the occurrence of a material and adverse change in Lineage’s business or financial condition that makes it impractical or inadvisable to market the shares or to enforce contracts for the sale of the shares.
Warrants
Lineage (previously Asterias) Warrants – Liability Classified
In March 2019, in connection with the closing of the Asterias Merger, Lineage assumed outstanding Asterias Warrants. As of December 31, 2020, the total number of common shares of Lineage subject to warrants that were assumed by Lineage in connection with the Asterias Merger was 1,089,900, which were converted to Lineage Warrants 30 days after the closing of the Asterias Merger, with similar terms and conditions retained under the Lineage Warrants as per the original Warrant Agreements. The Lineage Warrants have an exercise price of $6.15 per warrant share and expire on May 13, 2021.
Cell Cure Warrants – Liability Classified
Cell Cure has two sets of issued warrants (the “Cell Cure Warrants”). Warrants to purchase 24,566 Cell Cure ordinary shares at an exercise price of $40.5359 were issued to HBL in July 2017. These warrants expire in . Warrants to purchase 13,738 Cell Cure ordinary shares at exercise prices ranging from $32.02 to $40.02 per share were issued to consultants. of these warrants were cashless exercised in October 2020. The expense related to the cashless exercise was approximately $44,000 and it was recorded as other income/(expense), net on the statements of operations. The remaining 2,000 warrants have an exercise price of $40.00 and expire in .
ASC 815 requires freestanding financial instruments, such as warrants, with exercise prices denominated in currencies other than the functional currency of the issuer to be accounted for as liabilities at fair value, with all subsequent changes in fair value after the issuance date to be recorded as gains or losses in the consolidated statements of operations. Because the exercise price of the Cell Cure Warrants is U.S. dollar-denominated and settlement is not expected to occur in the next twelve months, Cell Cure classified the Cell Cure Warrants as a long-term liability in accordance with ASC 815.
The fair value of the Cell Cure Warrants at the time of issuance was determined by using the Black-Scholes option pricing model using the respective contractual term of the warrants. In applying this model, the fair value is determined by applying Level 3 inputs, as defined by ASC 820; these inputs are based on certain key assumptions including the fair value of the Cell Cure ordinary shares, adjusted for lack of marketability, as appropriate, and the expected stock price volatility over the term of the Cell Cure Warrants. The fair value of the Cell Cure ordinary shares is determined by Cell Cure’s Board of Directors, which may engage a valuation specialist to assist it in estimating the fair value, or may use recent transactions in Cell Cure shares, if any, as a reasonable approximation of fair value, or may apply other reasonable methods to determining the fair value, including a discount for lack of marketability. In connection with the cashless exercise in October 2020, Cell Cure had an independent third-party update the fair value of the Cell Cure shares. Lineage determines the stock price volatility using historical prices of comparable public company common stock for a period equal to the remaining term of the Cell Cure Warrants. The Cell Cure Warrants are revalued each reporting period using the same methodology described above, with changes in fair value included as gains or losses in other income and expenses, net, in the consolidated statements of operations.
| 95 |
For the years ended December 31, 2020 and 2019, Lineage recorded a noncash loss of $ million and a noncash gain of $ million, respectively, for the increase/decrease in the fair value of the Cell Cure Warrants included in other income and expenses, net for each period. The increase in the fair value of the Cell Cure Warrants was mainly attributable to an increase in the fair value of the Cell Cure shares due to additional progress made on the OpRegen program in 2020. As of December 31, 2020 and 2019, the Cell Cure Warrants, valued at $ million and $ million, respectively, were included in long-term liabilities on the consolidated balance sheets.
Equity Incentive Plan Awards
Effective November 8, 2019, Lineage adopted an amendment changing the name of the BioTime, Inc. 2012 Equity Incentive 2012 Plan to the Lineage Cell Therapeutics, Inc. 2012 Equity Incentive Plan (the “2012 Plan”). The 2012 Plan provides for the grant of stock options, restricted stock, restricted stock units (“RSUs”) and stock appreciation rights. As of December 31, 2020, a maximum of common shares were available for grant under the 2012 Plan. Recipients of stock options are eligible to purchase common shares at an exercise price equal to the fair market value of such shares on the date of grant. The maximum term of options granted under the 2012 Plan is years. Stock options generally vest over a four-year period based on continuous service; however, the 2012 Plan allows for other vesting periods. Upon the expiration of the restrictions applicable to an RSU, Lineage will either issue to the recipient, without charge, one common share per RSU or cash in an amount equal to the fair market value of one common share. RSUs granted from the 2012 Plan reduce the shares available for grant by two shares for each RSU granted.
Shares Available for Grant | Number of Options Outstanding | Number of RSUs Outstanding | Weighted Average Exercise Price | |||||||||||||
| December 31, 2018 | 1,885 | 13,867 | 402 | $ | 2.44 | |||||||||||
| Adjustment due to the AgeX Distribution | 117 | (2 | ) | 3 | ||||||||||||
| Increase to the 2012 Plan | 8,000 | |||||||||||||||
| Options granted | (3,581 | ) | 3,581 | 1.06 | ||||||||||||
| Options forfeited | 2,736 | (2,736 | ) | 2.13 | ||||||||||||
| Restricted stock units vested | (239 | ) | ||||||||||||||
| December 31, 2019 | 9,157 | 14,710 | 166 | $ | 2.17 | |||||||||||
| Options granted | (5,256 | ) | 5,256 | 0.71 | ||||||||||||
| Options forfeited | 4,101 | (4,101 | ) | 2.61 | ||||||||||||
| Restricted units vested | (73 | ) | ||||||||||||||
| December 31, 2020 | 8,002 | 15,865 | 93 | $ | 1.57 | |||||||||||
| Options exercisable at December 31, 2020 | 8,341 | $ | 2.16 | |||||||||||||
As of December 31, 2020, options outstanding and options exercisable under the 2012 Plan have a weighted-average remaining contractual term of years and years, respectively, and intrinsic value of $ million and $ million, respectively.
In connection with the vested RSUs during the year ended December 31, 2020, Lineage paid $ in minimum employee withholding taxes in exchange for vested Lineage common shares issuable to the employees and immediately retired those shares. For the year ended December 31, 2020, Lineage recorded a noncash stock-based compensation expense of $ million, in connection with the vested RSUs, included in consolidated stock-based compensation expense.
| 96 |
In connection with the vested RSUs during the year ended December 31, 2019, Lineage paid $ million in minimum employee withholding taxes in exchange for vested Lineage common shares issuable to the employees and immediately retired those shares. For the year ended December 31, 2019, Lineage recorded a noncash stock-based compensation expense of $ million, in connection with the vested RSUs, included in consolidated stock-based compensation expense.
At the effective time of the Asterias Merger, Lineage assumed sponsorship of the Asterias 2013 Equity Incentive Plan (the “Asterias Equity Plan”), with references to Asterias and Asterias common stock therein to be deemed references to Lineage and Lineage common shares. There were shares available under the Asterias Equity Plan immediately before the closing of the Asterias Merger, which became shares immediately following the Asterias Merger. The shares available under the Asterias Equity Plan will be for awards granted to those former Asterias employees who continued as Lineage employees upon consummation of the Asterias Merger.
Shares Available for Grant | Number of Options Outstanding | Number of RSUs Outstanding | Weighted Average Exercise Price | |||||||||||||
| March 8, 2019 | 5,190 | $ | ||||||||||||||
| Options granted | (490 | ) | 490 | 1.59 | ||||||||||||
| Options forfeited | 140 | (140 | ) | 1.63 | ||||||||||||
| December 31, 2019 | 4,840 | 350 | $ | 1.57 | ||||||||||||
| Options granted | ||||||||||||||||
| Options forfeited | ||||||||||||||||
| December 31, 2020 | 4,840 | 350 | $ | 1.57 | ||||||||||||
| Options exercisable at December 31, 2020 | 153 | $ | 1.57 | |||||||||||||
As of December 31, 2020, options outstanding and options exercisable under the Asterias Equity Plan both have a weighted-average remaining contractual term of years and intrinsic value of $ and $, respectively.
Stock-based compensation expense
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Expected life (in years) | 6.2 | 6.0 | ||||||
| Risk-free interest rates | 0.8% | 2.2% | ||||||
| Volatility | 67.7% | 63.1% | ||||||
| Dividend yield | % | % | ||||||
The weighted-average estimated fair value of stock options granted under the 2012 Plan and other stock option awards granted outside of the 2012 Plan, during the years ended December 31, 2020 and 2019 was $ and $ per share, respectively.
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Research and development | $ | 464 | $ | 516 | ||||
| General and administrative | 1,763 | 3,064 | ||||||
| Total stock-based compensation expense | $ | 2,227 | $ | 3,580 | ||||
| 97 |
The expense related to shares of Asterias restricted stock unit awards that immediately vested on the closing of the Asterias Merger and converted into the right to receive common shares of Lineage based on the Merger Exchange Ratio, resulting in common shares of Lineage issued on March 8, 2019, was included in stock-based compensation expense for the year ended December 31, 2019. The expense was not included as part of the purchase price of the Asterias Merger because these awards were principally attributable to post-combination services.
As of December 31, 2020, total unrecognized compensation costs related to unvested stock options under Lineage’s 2012 Plan was $ million, which is expected to be recognized as expense over a weighted average period of approximately years.
13. Income Taxes
For the year ended December 31, 2020, Lineage recorded a $1.2 million deferred tax benefit for income taxes.
For the year ended December 31, 2019, Lineage recorded a $7.4 million valuation allowance release and corresponding benefit for income taxes. This was comprised of a federal and state deferred income tax benefit of $3.6 million and $3.8 million, respectively, for the year ended December 31, 2019 due to the indefinite lived assets generated in the period and the release of the valuation allowance. The Company also recorded a current foreign income tax expense of $31,000 for the year ended December 31, 2019.
The domestic and foreign breakout of loss before net income tax benefit was as follows:
| December 31, | ||||||||
| 2020 | 2019 | |||||||
| Domestic | $ | (17,500 | ) | (7,303 | ) | |||
| Foreign | (4,424 | ) | (11,931 | ) | ||||
| Loss before net income tax benefit | $ | (21,924 | ) | (19,234 | ) | |||
Income taxes differed from the amounts computed by applying the indicated current U.S. federal income tax rate to pretax losses from operations as a result of the following:
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Computed tax benefit at federal statutory rate | 21% | 21% | ||||||
| Research and development and other credits | 1% | 3% | ||||||
| Removal of DTL for equity investment in Asterias due to merger | -% | 22% | ||||||
| Permanent differences | -% | (1)% | ||||||
| Change in valuation allowance | (17)% | (106)% | ||||||
| Establish DTL for deferred assets from Asterias Merger | -% | 42% | ||||||
| Deconsolidation of AgeX and subsidiaries net deferred tax assets | -% | 3% | ||||||
| State tax benefit, net of effect on federal income taxes | 3% | 54% | ||||||
| Foreign rate differential and other | (2)% | 1% | ||||||
| Income tax benefit | 6% | 39% | ||||||
| 98 |
The primary components of the deferred tax assets and liabilities at December 31, 2020 and 2019 were as follows (in thousands):
| December 31, | ||||||||
| Deferred tax assets/(liabilities): | 2020 | 2019 | ||||||
| Net operating loss carryforwards | $ | 63,941 | $ | 62,060 | ||||
| Research and development and other credits | 8,878 | 8,619 | ||||||
| Patents and licenses | 1,178 | 1,220 | ||||||
| Stock options | 2,131 | 2,708 | ||||||
| Operating lease liability | 242 | 832 | ||||||
| Operating lease ROU assets | (215 | ) | (775 | ) | ||||
| Equity method investments and marketable securities at fair value | (15,685 | ) | (19,367 | ) | ||||
| Other, net | 1,523 | 984 | ||||||
| Total | 61,993 | 56,281 | ||||||
| Valuation allowance | (64,069 | ) | (59,596 | ) | ||||
| Net deferred tax liabilities | $ | (2,076 | ) | $ | (3,315 | ) | ||
A valuation allowance is provided when it is more likely than not that some portion of the deferred tax assets will not be realized. Lineage established a full valuation allowance as of December 31, 2018 due to the uncertainty of realizing future tax benefits from its net operating loss carryforwards and other deferred tax assets, including foreign net operating losses generated by its subsidiaries. During the year ended December 31, 2019, a portion of the valuation allowance was released as it relates to Lineage’s indefinite lived assets that can be used against the indefinite lived liabilities. The amount of the valuation allowance released was $7.4 million; as new indefinite lived deferred tax assets are generated, we will continue to book provision benefits until the deferred tax liability position is exhausted, barring any new developments.
As of December 31, 2020, Lineage has gross net operating loss carryforwards of approximately $169.9 million for federal purposes. As of December 31, 2020, Lineage’s foreign subsidiaries have net operating loss carryforwards of approximately $88.3 million which carryforward indefinitely.
As of December 31, 2020, Lineage has net operating losses of $118.6 million for state tax purposes.
As of December 31, 2020, Lineage has research tax credit carryforwards for federal and state tax purposes of $3.2 million and $5.7 million, respectively. These tax credits reflect the amounts for Lineage, Asterias and OrthoCyte as of December 31, 2020. With the announcement of the California agreement with the Senate, allowing a deduction of up to $150,000 for expenses paid with the PPP loan proceeds, any amount disallowed for California, that relates to R&D wages, may be limited. For federal purposes, the credits generated each year have a carryforward period of 20 years. The federal tax credits expire in varying amounts between 2020 and 2040, while the state tax credits have no expiration period.
On August 5, 2020, Lineage began the liquidation of its foreign subsidiary BioTime Asia. At the time of the liquidation, BioTime Asia had an intercompany payable due to Lineage. For book purposes, the corresponding balances eliminate in consolidation. For federal purposes, the activities of their foreign subsidiaries are not included in the consolidated tax return. Accordingly, the payable was written off for tax purposes by Lineage, creating a $3.6 million bad debt deduction increasing its NOL carryover. For California, the activities of its foreign subsidiaries, including BioTime Asia, are included in the combined tax return. As such, the corresponding intercompany balances are eliminated.
Other Transactions and Related Impact on Income Taxes
The market value of the respective shares Lineage holds in OncoCyte, AgeX and Asterias (through the merger date of March 8, 2019) creates a deferred tax liability to Lineage based on the closing price of the security, less the tax basis of the security Lineage has in such shares. The deferred tax liability generated by shares that Lineage holds as of December 31, 2020 and 2019, is a source of future taxable income to Lineage, as prescribed by ASC 740-10-30-17, that will more likely than not result in the realization of its deferred tax assets to the extent of those deferred tax liabilities. This deferred tax liability is determined based on the closing price of those securities as of December 31, 2020 and 2019.
| 99 |
Other Income Tax Matters
Internal Revenue Code Section 382 places a limitation (“Section 382 Limitation”) on the amount of taxable income that can be offset by NOL carryforwards after a change in control (generally greater than 50% change in ownership within a three-year period) of a loss corporation. California has similar rules. Generally, after a change in control, a loss corporation cannot deduct NOL carryforwards in excess of the Section 382 Limitation. Due to these “change in ownership” provisions, utilization of the NOL and tax credit carryforwards may be subject to an annual limitation regarding their utilization against taxable income in future periods.
Lineage files a U.S. federal income tax return as well as various state and foreign income tax returns. In general, Lineage is no longer subject to tax examination by major taxing authorities for years before 2016. Although the statute is closed for purposes of assessing additional income and tax in these years, the taxing authorities may still make adjustments to the NOL and credit carryforwards used in open years. Therefore, the statute should be considered open as it relates to the NOL and credit carryforwards used in open years.
Lineage may be subject to potential examination by U.S. federal, U.S. states or foreign jurisdiction authorities in the areas of income taxes. These potential examinations may include questioning the timing and amount of deductions, the nexus of income among various tax jurisdictions and compliance with U.S. federal, U.S. state and foreign tax laws. Lineage’s management does not expect that the total amount of unrecognized tax benefits will materially change over the next twelve months.
Lineage’s practice is to recognize interest and penalties related to income tax matters in tax expense. As of December 31, 2020 and 2019, Lineage has no accrued interest and penalties.
14. Commitments and Contingencies
Carlsbad Lease
In May 2019, Lineage entered into a lease for approximately 8,841 square feet of rentable space in an office park in Carlsbad, California (the “Carlsbad Lease”). The term of the Carlsbad Lease commenced on August 1, 2019 and expires on October 31, 2022.
Base rent under the Carlsbad Lease as of August 1, 2020 is $18,386 per month and will increase by 3% annually on every August 1 thereafter during the lease term. Base rent for the first twenty-four months of the lease is based upon a deemed rentable area of 7,000 square feet. Base rent is abated for months two through five of the lease.
In addition to base rent, Lineage will pay a pro rata portion of increases in certain expenses, including real property taxes, utilities (to the extent not separately metered to the leased space) and the landlord’s operating expenses, over the amounts of those expenses incurred by the landlord. As security for the performance of its obligations under the Carlsbad Lease, Lineage provided the landlord with a security deposit of $17,850.
Alameda Leases and Alameda Sublease
In December 2015, Lineage entered into leases of office and laboratory space located in two buildings in Alameda, California (the “Alameda Leases”) comprised of 22,303 square feet (the “1010 Atlantic Premises”) and 8,492 square feet (the “1020 Atlantic Premises”). Base rent under the Alameda Leases beginning on February 1, 2020 was $72,636 per month with annual increases of approximately 3%. In addition to base rent, Lineage paid a pro rata portion of increases in certain expenses, including real property taxes, utilities (to the extent not separately metered to the leased space) and the landlord’s operating expenses, over the amounts of those expenses incurred by the landlord. As security for its obligations, Lineage provided the landlord with a security deposit of approximately $424,000, which was reduced to $78,000 on January 24, 2019 in accordance with the terms of the lease. The security deposit amount is considered restricted cash and is included in prepaid expenses and other current assets as of December 31, 2020 (See Note 2).
| 100 |
In April 2020, Lineage entered into a sublease with Industrial Microbes, Inc. (“Industrial Microbes”) for the use of 10,000 square feet in the 1010 Atlantic Premises (the “Industrial Microbes Sublease”). Base rent under the Industrial Microbes Sublease was $28,000 per month with annual increases of approximately 3%. Base rent for the first month was abated. In addition to base rent and utilities, Industrial Microbes paid a pro-rata portion of increases in operating expenses, after an abatement period of one year.
On September 11, 2020, Lineage entered into a Lease Termination Agreement with the landlord terminating the Alameda Leases effective as of August 31, 2020 for the 1020 Atlantic Premises and September 30, 2020 for the 1010 Atlantic Premises. In consideration for the termination of the leases, Lineage paid a termination fee of $130,000 and other amounts due under the terms of the Alameda Leases through the applicable effective termination dates, except that no rent was due with respect to the 1020 Atlantic Premises after July 31, 2020. Lineage’s security deposit is expected to be returned to Lineage by March 31, 2021. Lineage paid a separate termination fee of $30,000 to Industrial Microbes in connection with the termination of the Industrial Microbes Sublease and returned the $56,000 security deposit paid by Industrial Microbes. For the period of sublease from mid-April 2020 through September 2020, Lineage received $119,000 in rental income from Industrial Microbes.
Lineage will continue to occupy approximately 2,432 square feet of the 1010 Atlantic Premises under a new sublease agreement (the “Alameda Sublease”). The term of the Alameda Sublease is from October 1, 2020 through January 31, 2023. Base rent under the Alameda Sublease is $14,592 per month with annual increases of 3% each October 1 thereafter during the lease term. Base rent for the first month was abated. Lineage paid a security deposit of $16,000 under the Alameda Sublease; this amount is considered restricted cash and is included in deposits and other long-term assets as of December 31, 2020 (see Note 2).
Based on the smaller footprint, and after taking into consideration the fees disclosed above, Lineage has reduced its contractual obligations by approximately $780,000 over the remaining life of the original leases through January 31, 2023.
New York Leased Office Space
Lineage currently pays $5,050 per month for the use of approximately 900 square feet of office space in New York City, which is made available to Lineage for use in conducting meetings and other business affairs, on a month-by-month basis, by one of its directors at an amount that approximates his cost. This lease was not in the scope of ASC 842 because it is a month to month lease (see Note 2). These payments are expected to cease in March 2021 when the office space lease expires.
Cell Cure Leases
Cell Cure leases 728.5 square meters (approximately 7,842 square feet) of office and laboratory space in Jerusalem, Israel under a lease that expires December 31, 2025, with an option to extend the lease for 5 years (the “Original Cell Cure Lease”). Base monthly rent is NIS 39,776 (approximately US $12,200 per month using the December 7, 2020 exchange rate). In addition to base rent, Cell Cure pays a pro rata share of real property taxes and certain costs related to the operation and maintenance of the building in which the leased premises are located.
On January 28, 2018, Cell Cure entered into another lease agreement for an additional 934 square meters (approximately 10,054 square feet) of office space in the same facility in Jerusalem, Israel under a lease that expires on December 31, 2025, with two options to extend the lease for 5 years each (the “January 2018 Lease”). The January 2018 Lease commenced on April 1, 2018 and included a leasehold improvement construction allowance of up to NIS 4,000,000 (approximately up to $1.1 million using the December 31, 2018 exchange rate) from the landlord. The leasehold improvements were completed in December 2018 and the entire allowance was used. Beginning on January 1, 2019, combined base rent and construction allowance payments for the January 2018 Lease are NIS 93,827 per month (approximately $26,000 per month).
| 101 |
Prior to the adoption of ASC 842 on January 1, 2019, Cell Cure was considered the owner of the tenant improvements under construction under ASC 840-40-55 as Cell Cure, among other things, had the primary obligation to pay for construction costs and Cell Cure retains exclusive use of the leased facilities for its office, research and cGMP manufacturing facility requirements after construction was completed (“build to suit” lease). In accordance with the ASC 840 guidance, amounts expended by Cell Cure for construction was reported as construction in progress, and the proceeds received from the landlord, if any, are reported as a lease liability. As of December 31, 2018, approximately $1.1 million under the January 2018 Lease was incurred and recorded as leasehold improvement construction in progress, with a corresponding amount included in long term lease liability representing the full amount utilized from the landlord’s leasehold improvement construction allowance. By March 2019, the landlord paid the complete leasehold improvement construction allowance and the property was placed in service.
See Note 2 for discussion of the impact of adoption of ASC 842 on January 1, 2019, and below for the ROU assets and liabilities recorded in connection with the adoption of ASC 842 as of, and during the year ended December 31, 2019 for the Original Cell Cure Lease and January 2018 Lease (the “Cell Cure Leases”).
In December 2018, Cell Cure made a deposit required under the January 2018 Lease, which amount of $420,000 is included in deposits and other long-term assets on the consolidated balance sheet as of December 31, 2020, to be held as restricted cash during the term of the January 2018 Lease.
Adoption of ASC 842
The below tables provide the amounts recorded in connection with the adoption of ASC 842 as of, and for the years ended December 31, 2020 and 2019, for Lineage’s operating and financing leases, as applicable.
Supplemental cash flow information related to leases was as follows (in thousands):
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Cash paid for amounts included in the measurement of lease liabilities: | ||||||||
| Operating cash flows from operating leases | $ | 1,356 | $ | 1,364 | ||||
| Operating cash flows from financing leases | 20 | 28 | ||||||
| Financing cash flows from financing leases | 26 | 30 | ||||||
| Right-of-use assets obtained in exchange for lease obligations: | ||||||||
| Operating leases | 1,047 | 738 | ||||||
| Financing leases | ||||||||
Supplemental balance sheet information related to leases was as follows (in thousands, except lease term and discount rate):
| December 31, | ||||||||
| 2020 | 2019 | |||||||
| Operating leases | ||||||||
| Right-of-use assets, net | $ | 2,916 | $ | 4,666 | ||||
| Right-of-use lease liabilities, current | $ | 746 | $ | 1,190 | ||||
| Right-of-use lease liabilities, noncurrent | 2,514 | 3,868 | ||||||
| Total operating lease liabilities | $ | 3,260 | $ | 5,058 | ||||
| Financing leases | ||||||||
| Property and equipment, gross | $ | 79 | $ | 96 | ||||
| Accumulated depreciation | (65 | ) | (48 | ) | ||||
| Property and equipment, net | $ | 14 | $ | 48 | ||||
| Current liabilities | $ | 16 | $ | 33 | ||||
| Long-term liabilities | 26 | 77 | ||||||
| Total finance lease liabilities | $ | 42 | $ | 110 | ||||
| Weighted average remaining lease term | ||||||||
| Operating leases | 4.2 years | 4.1 years | ||||||
| Finance leases | 2.4 years | 3.4 years | ||||||
| Weighted average discount rate | ||||||||
| Operating leases | 8.0 | % | 9.1 | % | ||||
| Finance leases | 10.0 | % | 10.0 | % | ||||
| 102 |
Future minimum lease commitments are as follows (in thousands):
| Operating Leases | Finance Leases | |||||||
| Year Ending December 31, | ||||||||
| 2021 | $ | 957 | $ | 19 | ||||
| 2022 | 912 | 19 | ||||||
| 2023 | 480 | 8 | ||||||
| 2024 | 454 | |||||||
| 2025 | 442 | |||||||
| Thereafter | 628 | |||||||
| Total lease payments | $ | 3,873 | $ | 46 | ||||
| Less imputed interest | (613 | ) | (4 | ) | ||||
| Total | $ | 3,260 | $ | 42 | ||||
Research and Option Agreement
On January 5, 2019, Lineage and Orbit Biomedical Limited (“Orbit”) entered into a Research and Option Agreement, which was assigned by Orbit to Gyroscope Therapeutics, Limited (“Gyroscope”) and amended on May 7, 2019, January 30, 2020, May 1, 2020 and September 4, 2020 (the “Gyroscope Agreement”). As amended, the Gyroscope Agreement provides Lineage access to Gyroscope’s vitrectomy-free subretinal injection device (the “Orbit Device”) as a means of delivering OpRegen in Lineage’s ongoing Phase 1/2a clinical trial through the earlier of: (i) December 1, 2020; or (ii) or treatment of three additional patients with the Orbit Device between September 4, 2020 and December 1, 2020 (the “Access Period”). Following the Access Period, Lineage also has an exclusive right to negotiate a definitive agreement to distribute and sell the Orbit Device for the subretinal delivery of RPE cells for the treatment of dry AMD, which was extended through May 2021 (the “Option Period”). Pursuant to the terms of the Gyroscope Agreement, Lineage paid access fees totaling $2.5 million: (i) $1.25 million in January 2019 upon execution of the Gyroscope Agreement; and (ii) $1.25 million in August 2019 upon completion of certain collaborative research activities using the Gyroscope technology for the OpRegen Phase 1/2a clinical trial. These access fees of $2.5 million were amortized on a straight-line basis throughout 2019 and included in research and development expenses. Lineage also agreed to reimburse Gyroscope for costs of consumables, training services, travel costs and other out of pocket expenses incurred by Gyroscope for performing services under the Gyroscope Agreement. In January 2020, Lineage agreed to pay an additional $0.5 million to extend the Access Period to July 5, 2020, $0.2 million of which was paid in February 2020 and $0.3 million of which was paid in November 2020. The Access Period was subsequently extended at no cost as described above. In February 2021, Lineage paid $0.5 million to extend the Option Period.
Litigation – General
Lineage is subject to various claims and contingencies in the ordinary course of its business, including those related to litigation, business transactions, employee-related matters, and others. When Lineage is aware of a claim or potential claim, it assesses the likelihood of any loss or exposure. If it is probable that a loss will result and the amount of the loss can be reasonably estimated, Lineage will record a liability for the loss. If the loss is not probable or the amount of the loss cannot be reasonably estimated, Lineage discloses the claim if the likelihood of a potential loss is reasonably possible and the amount involved could be material. Lineage is not aware of any claims likely to have a material adverse effect on its financial condition or results of operations.
| 103 |
On February 19, 2019, a putative shareholder class action lawsuit was filed (captioned Lampe v. Asterias Biotherapeutics, Inc. et al., Case No. RG19007391) in the Superior Court of the State of California, County of Alameda challenging the Asterias Merger. On March 1, 2019, Asterias made certain amendments and supplements to its public disclosures regarding the Asterias Merger (the “Supplemental Disclosures”). On May 3, 2019, an amended class action complaint (the “Amended Complaint”) was filed. The Amended Complaint named Lineage, Patrick Merger Sub, Inc., the Asterias board of directors, one member of Lineage’s board of directors, and certain stockholders of both Lineage and Asterias. The action was brought by two purported stockholders of Asterias, on behalf of a putative class of Asterias stockholders, and asserted breach of fiduciary duty and aiding and abetting claims under Delaware law. The Amended Complaint alleged, among other things, that the process leading up to the Asterias Merger was conflicted and inadequate, and that the proxy statement filed by Asterias with the Commission omitted certain material information, which allegedly rendered the information disclosed materially misleading. The Amended Complaint sought, among other things, that a class be certified, the recovery of monetary damages, and attorneys’ fees and costs.
On June 3, 2019, defendants filed demurrers to the Amended Complaint. On August 13, 2019, the parties submitted a stipulation to the court seeking dismissal of the action with prejudice as to the named Plaintiffs and without prejudice as to the unnamed putative class members, and disclosing to the court the parties’ agreement to resolve, for $200,000, Plaintiffs’ claim for an award of attorneys’ fees and expenses in connection with the purported benefit conferred on Asterias stockholders by the Supplemental Disclosures. The court granted the stipulation and dismissed the action August 14, 2019. Lineage continues to believe that the claims and allegations in the action lack merit, but believed that it was in Lineage’s shareholders’ best interest for the action to be dismissed and to resolve the fee claim in a timely manner without additional costly litigation expenses.
On October 14, 2019, another putative class action lawsuit was filed challenging the Asterias Merger. This action (captioned Ross v. Lineage Cell Therapeutics, Inc., et al., C.A. No. 2019-0822) was filed in Delaware Chancery Court and names Lineage, the Asterias board of directors, one member of Lineage’s board of directors, and certain stockholders of both Lineage and Asterias as defendants. The action was brought by a purported stockholder of Asterias, on behalf of a putative class of Asterias stockholders, and asserts breach of fiduciary duty and aiding and abetting claims under Delaware law. The complaint alleges, among other things, that the process leading up to the Asterias Merger was conflicted, that the Asterias Merger consideration was inadequate, and that the proxy statement filed by Asterias with the Commission omitted certain material information, which allegedly rendered the information disclosed materially misleading. The complaint seeks, among other things, that a class be certified, the recovery of monetary damages, and attorneys’ fees and costs. On December 20, 2019, the defendants moved to dismiss the complaint. On February 10, 2020, the plaintiff filed an opposition. Defendants filed their replies on March 13, 2020. On June 23, 2020, a hearing on the motions to dismiss occurred. On September 21, 2020, the Chancery Court denied the motion to dismiss as to Lineage and certain members of the Asterias board of directors, and it granted the motion to dismiss as to all other defendants. On October 30, 2020, the remaining defendants filed an answer to the complaint.
Lineage believes the allegations in the action lack merit and intends to vigorously defend the claims asserted. It is impossible at this time to assess whether the outcome of this proceeding will have a material adverse effect on Lineage’s consolidated results of operations, cash flows or financial position. Therefore, in accordance with ASC 450, Contingencies, Lineage has not recorded any accrual for a contingent liability associated with this legal proceeding based on its belief that a liability, while possible, is not probable nor estimable, and any range of potential contingent liability amounts cannot be reasonably estimated at this time. Lineage records legal expenses as incurred.
Employment Contracts
Lineage has entered into employment agreements with certain executive officers. Under the provisions of the agreements, Lineage may be required to incur severance obligations for matters relating to changes in control, as defined in the agreements, and involuntary terminations.
| 104 |
Indemnification
In the normal course of business, Lineage may provide indemnifications of varying scope under Lineage’s agreements with other companies or consultants, typically Lineage’s clinical research organizations, investigators, clinical sites, suppliers and others. Pursuant to these agreements, Lineage will generally agree to indemnify, hold harmless, and reimburse the indemnified parties for losses and expenses suffered or incurred by the indemnified parties arising from claims of third parties in connection with the use or testing of Lineage’s products and services. Indemnification provisions could also cover third party infringement claims with respect to patent rights, copyrights, or other intellectual property pertaining to Lineage products and services. The term of these indemnification agreements will generally continue in effect after the termination or expiration of the particular research, development, services, or license agreement to which they relate. The potential future payments Lineage could be required to make under these indemnification agreements will generally not be subject to any specified maximum amount. Historically, Lineage has not been subject to any claims or demands for indemnification. Lineage also maintains various liability insurance policies that limit Lineage’s financial exposure. As a result, Lineage believes the fair value of these indemnification agreements is minimal. Accordingly, Lineage has not recorded any liabilities for these agreements as of December 31, 2020 and 2019.
Second Amendment to Clinical Trial and Option Agreement and License Agreement with Cancer Research UK
On May 6, 2020, Lineage and its wholly owned subsidiary Asterias entered into a Second Amendment to Clinical Trial and Option Agreement (the “CTOA Amendment”) with Cancer Research UK (“CRUK”) and Cancer Research Technology Limited (“CRT”), which amends the Clinical Trial and Option Agreement entered into between Asterias, CRUK and CRT dated September 8, 2014, as amended September 8, 2014. Pursuant to the CTOA Amendment, Lineage assumed all obligations of Asterias and exercised early its option to acquire data generated in the Phase 1 clinical trial of VAC2 in non-small cell lung cancer being conducted by CRUK. CRUK will continue conducting the VAC2 study.
Lineage and CRT effectuated the option by simultaneously entering into a license agreement (the “License Agreement”) pursuant to which Lineage agreed to pay the previously agreed signature fee of £1,250,000 (approximately $1.6 million). In consideration of Lineage’s agreement to exercise the option prior to completion of the study, the parties agreed to defer the signature fee as follows: £500,000 in September 2020, £500,000 in January 2021 and £250,000 in April 2021. For the primary licensed product for the first indication, the License Agreement provides for milestone fees of up to £8,000,000 based upon initiation of a Phase 3 clinical trial and the filing for regulatory approval and up to £22,500,000 in sales-based milestones payments. Additional milestone fees and sales-based milestone payments would be payable for other products or indications, and mid-single-digit royalty payments are payable on sales of commercial products.
Either party may terminate the License Agreement for the uncured material breach of the other party. CRT may terminate the License Agreement in the case of Lineage’s insolvency or if Lineage ceases all development and commercialization of all products under the License Agreement.
Second Amended and Restated License Agreement
On June 15, 2017, Cell Cure entered into a Second Amended and Restated License Agreement (the “License Agreement”) with Hadasit Medical Research Services and Development Ltd. (“Hadasit”), the commercial arm and a wholly owned subsidiary of Hadassah Medical Organization. Pursuant to the License Agreement, Hadasit granted Cell Cure an exclusive, worldwide, royalty bearing license (with the right to grant sublicenses) in its intellectual property portfolio of materials and technology related to human stem cell derived photoreceptor cells and retinal pigment epithelial cells (the “Licensed IP”), to use, commercialize and exploit any part thereof, in any manner whatsoever in the fields of the development and exploitation of (i) human stem cell derived photoreceptor cells, solely for use in cell therapy for the diagnosis, amelioration, prevention and treatment of eye disorders, and (ii) human stem cell derived retinal pigment epithelial cells, solely for use in cell therapy for the diagnosis, amelioration, prevention and treatment of eye disorders.
As consideration for the Licensed IP, Cell Cure will pay a small one-time lump sum payment, a royalty in the mid-single digits of net sales from sales of Licensed IP by any invoicing entity, and a royalty of 21.5% of sublicensing receipts. In addition, Cell Cure will pay Hadasit an annual minimal non-refundable royalty, which will become due and payable the first January 1 following the completion of services to Cell Cure by a research laboratory.
| 105 |
Cell Cure will pay Hadasit non-refundable milestone payments upon the recruitment of the first patient for the first Phase 2b clinical trial, upon the enrollment of the first patient in the first Phase 3 clinical trials, upon delivery of the report for the first Phase 3 clinical trials, upon the receipt of an NDA or marketing approval in the European Union, whichever is the first to occur, and upon the first commercial sale in the United States or European Union, whichever is the first to occur. Such milestones, in the aggregate, may be up to $3.5 million. As of December 31, 2020, Cell Cure had not accrued any milestone payments under the License Agreement.
The License Agreement terminates upon the expiration of Cell Cure’s obligation to pay royalties for all licensed products, unless earlier terminated. In addition to customary termination rights of both parties, Hadasit may terminate the License Agreement if Cell Cure fails to continue the clinical development of the Licensed IP or fails to take actions to commercialize or sell the Licensed IP over any consecutive 12 month period. The License Agreement also contains mutual confidentiality obligations of Cell Cure and Hadasit, and indemnification obligations of Cell Cure.
Royalty obligations and license fees
Lineage and its subsidiaries or affiliates are parties to certain licensing agreements with research institutions, universities and other parties for the rights to use those licenses and other intellectual property in conducting research and development activities. These licensing agreements provide for the payment of royalties by Lineage or the applicable party to the agreement on future product sales, if any. In addition, in order to maintain these licenses and other rights during the product development, Lineage or the applicable party to the contract must comply with various conditions including the payment of patent related costs and annual minimum maintenance fees. Annual minimum maintenance fees are expected to be approximately $30,000 to $60,000 per year.
Grants
Under the terms of the grant agreement between Cell Cure and Israel Innovation Authority (“IIA”) (formerly the Office of the Chief Scientist of Israel) of the Ministry of Economy and Industry, for the development of OpRegen®, Cell Cure will be required to pay royalties on future product sales, if any, up to the amounts received from the IIA, plus interest indexed to LIBOR. Cell Cure’s research and product development activities under the grant are subject to substantial risks and uncertainties and performed on a best efforts basis. As a result, Cell Cure is not required to make any payments under the grant agreement unless it successfully commercializes OpRegen®. Accordingly, pursuant to ASC 730-20, the Cell Cure grant is considered a contract to perform research and development services for others and grant revenue is recognized as the related research and development expenses are incurred (see Note 2).
Israeli law pertaining to such government grants contain various conditions, including substantial penalties and restrictions on the transfer of intellectual property, or the manufacture, or both, of products developed under the grant outside of Israel, as defined by the IIA.
15. Employee Benefit Plan
We have a defined contribution 401(k) plan for all employees. Under the terms of the plan, employees may make voluntary contributions as a percentage or defined amount of compensation. We provide a safe harbor contribution of up to 5.0% of the employee’s compensation, not to exceed eligible limits, and subject to employee participation. For the years ended December 31, 2020 and 2019, we incurred approximately $149,000 and $287,000, respectively, in expenses related to the safe harbor contribution.
16. Segment Information
Lineage’s executive management team, as a group, represents the entity’s chief operating decision makers. Lineage’s executive management team views Lineage’s operations as one segment that includes the research and development of therapeutic products for retinal diseases, neurological diseases and disorders and oncology. As a result, the financial information disclosed materially represents all of the financial information related to Lineage’s sole operating segment.
| 106 |
17. Enterprise-Wide Disclosures
Geographic Area Information
The following table presents consolidated revenues, including license fees, royalties, grant income, and other revenues, disaggregated by geography, based on the billing addresses of customers, or in the case of grant revenues based on where the governmental entities that fund the grant are located (in thousands).
| Year Ended December 31, | ||||||||
| Geographic Area | 2020 | 2019 | ||||||
| United States | $ | 1,160 | $ | 2,092 | ||||
| Foreign (1) | 666 | 1,423 | ||||||
| Total revenues | $ | 1,826 | $ | 3,515 | ||||
| (1) |
The composition of Lineage’s long-lived assets, consisting of plant and equipment, net, between those in the United States and in foreign countries, as of December 31, 2020 and 2019, is set forth below (in thousands):
| December 31, | ||||||||
| 2020 | 2019 | |||||||
| Domestic | $ | 1,035 | $ | 3,654 | ||||
| Foreign (1) | 4,595 | 4,521 | ||||||
| Total | $ | 5,630 | $ | 8,175 | ||||
| (1) |
Major Sources of Revenues
The following table presents Lineage’s consolidated revenues disaggregated by source (in thousands).
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| REVENUES: | ||||||||
| Grant revenue | $ | 1,053 | $ | 2,037 | ||||
| Royalties from product sales and license fees | 773 | 1,221 | ||||||
| Sale of research products and services | 257 | |||||||
| Total revenues | $ | 1,826 | $ | 3,515 | ||||
Prepaid expenses and other current assets at December 31, 2020 includes $0.2 million of receivables related to royalties from product sales and license fees, and $ million of receivables related to cash in transit for sales of ATM Shares in 2020 that settled in 2021.
The following table shows Lineage’s major sources of revenues, as a percentage of total revenues, that were recognized during the years ended December 31, 2020 and 2019:
| Year Ended December 31, | ||||||||
| Sources of Revenues | 2020 | 2019 | ||||||
| NIH grant income | 21.2 | % | 17.5 | % | ||||
| IIA grant income (Cell Cure Neurosciences, Ltd, Israel) | 36.5 | % | 40.5 | % | ||||
| Royalties, licenses, subscriptions, advertising and other | 42.3 | % | 34.7 | % | ||||
| Sale of research products | % | 7.3 | % | |||||
| 107 |
18. Selected Quarterly Financial Information (UNAUDITED, in thousands, except per share data)
Lineage has derived this data from the unaudited consolidated interim financial statements that, in Lineage’ s opinion, have been prepared on substantially the same basis as the audited consolidated financial statements contained herein and include all normal recurring adjustments necessary for a fair presentation of the financial information for the periods presented. These unaudited consolidated quarterly results should be read in conjunction with the consolidated financial statements and notes thereto included herein. The consolidated operating results in any quarter are not necessarily indicative of the consolidated results that may be expected for any future period.
| Year Ended December 31, 2020 | First Quarter | Second Quarter | Third Quarter | Fourth Quarter | ||||||||||||
| Revenues, net | $ | 514 | 386 | 571 | 355 | |||||||||||
| Operating expenses | 7,858 | 6,713 | 7,194 | 6,123 | ||||||||||||
| Loss from operations | (7,438 | ) | (6,402 | ) | (6,725 | ) | (5,882 | ) | ||||||||
| Net income (loss) attributable to Lineage | (8,399 | ) | (6,522 | ) | (7,760 | ) | 2,032 | |||||||||
| Basic net income (loss) per share | $ | (0.06 | ) | $ | (0.04 | ) | $ | (0.05 | ) | $ | 0.01 | |||||
| Year Ended December 31, 2019 | ||||||||||||||||
| Revenues, net | $ | 928 | 779 | 567 | 1,241 | |||||||||||
| Operating expenses | 13,621 | 11,493 | 8,875 | 7,990 | ||||||||||||
| Loss from operations | (12,761 | ) | (10,821 | ) | (8,422 | ) | (6,872 | ) | ||||||||
| Net income (loss) attributable to Lineage | 39,310 | (30,032 | ) | (16,505 | ) | (4,482 | ) | |||||||||
| Basic net income (loss) per share | $ | 0.30 | $ | (0.20 | ) | $ | (0.11 | ) | $ | (0.03 | ) | |||||
Quarterly and year-to-date computations of net income (loss) per share amounts are calculated using the respective period weighted average shares outstanding. Therefore, the sum of the per share amounts for the quarters may not agree with the per share amounts for the year.
19. Subsequent Events
Sale of OncoCyte Shares
In January and February 2021, Lineage sold million shares of OncoCyte common stock for gross proceeds of $10.1 million. After these sales, Lineage owns shares of OncoCyte common stock, which has a value of $ million as of March 5, 2021.
Sales of Lineage Shares Under the ATM
In the first quarter of 2021 through March 5, 2021, Lineage sold common shares of Lineage ATM Shares for gross and net proceeds of $million and $million, respectively (in each case, which includes $million of proceeds in transit related to 2020 sales that settled in 2021). See Note 11 for additional information. On March 5, 2021, Lineage filed a prospectus supplement with the SEC in connection with the offer and sale of an additional $million of ATM Shares.
Research and Option Agreement
In February 2021, Lineage extended the Option Period with Gyroscope for $0.5 million for an additional three months. See Note 14 for additional information.
| 108 |
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
Not applicable.
| ITEM 9A. | CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
It is management’s responsibility to establish and maintain adequate internal control over all financial reporting pursuant to Rule 13a-15 under the Securities Exchange Act of 1934 (“Exchange Act”). Our management, including our principal executive officer and our principal financial officer, have reviewed and evaluated the effectiveness of our disclosure controls and procedures as of the end of our fourth quarter. Following this review and evaluation, management collectively determined that our disclosure controls and procedures are effective to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act: (i) is recorded, processed, summarized and reported within the time periods specified in Securities and Exchange Commission rules and forms; and (ii) is accumulated and communicated to management, including our chief executive officer and our chief financial officer, as appropriate to allow timely decisions regarding required disclosure.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the period covered by this Report that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting, as defined in Exchange Act Rule 13a-15(f), is a process designed by, or under the supervision of, our principal executive officer, our principal operations officer, and our principal financial officer, and effected by our Board of Directors, management, and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
| ● | Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; | |
| ● | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and | |
| ● | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation. The scope of management’s assessment of the effectiveness of internal control over financial reporting includes our consolidated subsidiaries.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2020, based on criteria established in the 2013 Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this assessment, management believes that, as of that date, our internal control over financial reporting was effective.
| ITEM 9B. | OTHER INFORMATION |
Not applicable
| 109 |
PART III
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS, AND CORPORATE GOVERNANCE |
Board of Directors
Set forth below are the names, ages, board committee assignments, tenure, and certain biographical information of each of the members of our Board of Directors (our “Board”) as of March 11, 2021.
| Name | Age | Committees | Director Since | |||
| Alfred D. Kingsley | 77 | Financial Strategy* | July 2009 | |||
| Deborah Andrews | 63 | Audit*, Compensation, Nominating & Corporate Governance | April 2014 | |||
| Don M. Bailey | 75 | Nominating & Corporate Governance*, Financial Strategy | March 2019 | |||
| Neal C. Bradsher, CFA | 55 | Nominating & Corporate Governance, Financial Strategy | July 2009 | |||
| Brian M. Culley | 49 | None | September 2018 | |||
| Michael H. Mulroy | 54 | Compensation*, Audit, Nominating & Corporate Governance, Financial Strategy | October 2014 | |||
| Angus C. Russell | 65 | Audit, Compensation | December 2014 |
* Committee chairperson
Alfred D. Kingsley. Mr. Kingsley has been Chairman of the Board since July 2009. Mr. Kingsley has been general partner of Greenway Partners, L.P., a private investment firm, and President of Greenbelt Corp., a business consulting firm, since 1993. Greenbelt served as our financial advisor from 1998 until 2009. Mr. Kingsley also serves as a director of OncoCyte Corporation (OCX), a clinical-stage diagnostics company focused on novel, non-invasive blood-based tests for the early detection of cancer. From January 2017 to October 2018, Mr. Kingsley served as Executive Chairman of AgeX Therapeutics, Inc. (AGE), a biotechnology company focused on the development and commercialization of novel therapeutics targeting human aging. Mr. Kingsley also served as a director of Asterias Biotherapeutics, Inc. (AST) from 2012 until our acquisition of Asterias in March 2019. Mr. Kingsley was Senior Vice-President of Icahn and Company and its affiliated entities for more than 25 years. Mr. Kingsley holds a B.S. degree in economics from the Wharton School of the University of Pennsylvania and a J.D. degree and LLM in taxation from New York University Law School. Mr. Kingsley’s long career in corporate finance and mergers and acquisitions includes substantial experience in helping companies to improve their management and corporate governance, and to restructure their operations. Mr. Kingsley developed an intimate knowledge of our business in his role as our financial advisor before he joined our Board. Mr. Kingsley has been instrumental in structuring our equity and debt financings, and in the transition of our business focus into the field of stem cell technology, and the business acquisitions that have helped us expand the scope of our business.
Deborah Andrews. Ms. Andrews served as Chief Financial Officer of STAAR Surgical Company (STAA), a leader in the development, manufacture, and marketing of minimally invasive ophthalmic products employing proprietary technologies, from September 2017 until June 30, 2020 after serving as Vice President, Chief Accounting Officer since 2013. Ms. Andrews also served as STAAR Surgical’s Vice President, Chief Financial Officer from 2005 to 2013, as its Global Controller from 2001 to 2005, and as its Vice President, International Finance from 1999 to 2001. Ms. Andrews previously worked as a senior accountant for a major public accounting firm. Ms. Andrews holds a B.S. degree in accounting from California State University at San Bernardino. Ms. Andrews brings to our Board significant experience in finance, financial reporting, accounting, and auditing, and in management as a senior financial and accounting executive of a public medical device company during a period of significant growth.
| 110 |
Don M. Bailey. Mr. Bailey previously served as a director and Chairman of Asterias Biotherapeutics, Inc. (AST) from February 2016 until our acquisition of Asterias in March 2019. Mr. Bailey served as President and Chief Executive Officer of Questcor Pharmaceuticals, Inc. (QCOR), a biopharmaceutical company focused on the treatment of patients with serious, difficult-to-treat autoimmune and inflammatory disorders, from 2007 until Questcor was acquired by Mallinckrodt plc (MNK) in 2014. He was also a director of Mallinckrodt plc from August 2014 to March 2016, and during this time he was the Chairman of its portfolio committee. He initially joined the Questcor board of directors in 2006 as an independent director and Chairman of its audit committee. From August 2016 to November 2017, Mr. Bailey served as a director of OncoCyte Corporation (OCX). From June 2015 until its acquisition by Acorda Therapeutics, Inc. (ACOR) in May 2016, Mr. Bailey was also an independent director and chairman of the audit committee of Biotie Therapeutics Corp. (BITI), a clinical-stage pharmaceutical company headquartered in Turku, Finland. Mr. Bailey was an independent director and the non-executive chairman of the board of directors of STAAR Surgical Company (STAA), a leader in the development, manufacture, and marketing of minimally invasive ophthalmic products employing proprietary technologies, from 2005 until 2014. Mr. Bailey served on its audit committee and was chair of its nominating and corporate governance committee. Mr. Bailey was the chairman of the board of directors of Comarco, Inc. (CMRO), a defense services company transformed into a wireless communication products company, from 1998 until 2007, where he served as Chief Executive Officer from 1991 until 2000. Mr. Bailey holds a B.S. degree in mechanical engineering from the Drexel Institute of Technology, an M.S. degree in operations research from the University of Southern California and an M.B.A. from Pepperdine University. Mr. Bailey has also served as a board member on several non-profit and academic enterprises. Mr. Bailey is a founding board member of the University of California Irvine’s (UCI) Applied Innovation Institute. Mr. Bailey brings to our Board significant knowledge of the pharmaceuticals industry and extensive experience as an executive and board member of publicly traded pharmaceutical companies.
Neal C. Bradsher, CFA. Mr. Bradsher has been President of Broadwood Capital, Inc., a private investment firm, since 2002. Mr. Bradsher holds a B.A. degree in economics from Yale College and is a Chartered Financial Analyst. Mr. Bradsher was a director of Questcor Pharmaceuticals, Inc. (QCOR), from 2004 until Questcor was acquired by Mallinckrodt plc (MNK) in 2014. Mr. Bradsher brings to our Board a wealth of experience in finance, management and corporate governance attained through his investments in other companies, including companies in the pharmaceutical, biotechnology, medical device, medical diagnostics, health care services and health care information systems sectors. He has worked with several health care companies to improve their management and governance. Entities that Mr. Bradsher controls have invested in most of Lineage’s financing transactions over the last several years. Mr. Bradsher is the president of the general partner of Broadwood Partners, L.P., currently our largest shareholder.
Brian M. Culley. Mr. Culley joined Lineage as Chief Executive Officer in September 2018 and was appointed as Interim Chief Financial Officer in January 2021. Prior to joining Lineage, Mr. Culley served from August 2017 to September 2018 as interim Chief Executive Officer at Artemis Therapeutics, Inc. (ATMS). Mr. Culley previously served as Chief Executive Officer of Mast Therapeutics, Inc. (MSTX), from 2010, and was also a member of its board of directors from 2011, until Mast’s merger with Savara, Inc. (SVRA) in April 2017. Mr. Culley served from 2007 to 2010 as Mast’s Chief Business Officer and Senior Vice President, from 2006 to 2007 as Mast’s Senior Vice President, Business Development, and from 2004 to 2006 as Mast’s Vice President, Business Development. From 2002 until 2004, Mr. Culley was Director of Business Development and Marketing for Immusol, Inc. From 1999 until 2000, he worked at the University of California, San Diego (UCSD) Department of Technology Transfer & Intellectual Property Services and from 1996 to 1999 he conducted drug development research for Neurocrine Biosciences, Inc. (NBIX). Mr. Culley has also served on the Board of Orphagen Pharmaceuticals, Inc. since May 2017. Mr. Culley has more than 25 years of business and scientific experience in the life sciences industry. He received a B.S. in biology from Boston College, a masters in biochemistry and molecular biology from the University of California, Santa Barbara, and an M.B.A. from The Johnson School of Business at Cornell University. Mr. Culley brings to our Board significant knowledge of the biotechnology industry and extensive experience as an executive and board member of publicly traded pharmaceutical companies.
Michael H. Mulroy. Mr. Mulroy served as the Chief Executive Officer and a member of the board of directors of Asterias Biotherapeutics, Inc. (AST) from June 2017 until our acquisition of Asterias in March 2019. In April 2020, Mr. Mulroy joined Magtrol Inc., a leading manufacturer of motor test equipment and hysteresis brakes and clutches, on a part time basis, where he also serves on its board of directors. Prior to joining Asterias, Mr. Mulroy served as a Senior Advisor to CamberView Partners, LLC (now part of PJT Partners Inc.), which assists companies in connection with investor engagement and complex corporate governance issues. Prior to its sale in 2014, Mr. Mulroy served as Executive Vice President, Strategic Affairs and General Counsel and Corporate Secretary of Questcor Pharmaceuticals, Inc. (QCOR). Mr. Mulroy joined Questcor in 2011 as Chief Financial Officer, General Counsel and Corporate Secretary. From 2003 to 2011, Mr. Mulroy was employed by the law firm of Stradling Yocca Carlson & Rauth, where he served as a partner from 2004. From 1997 to 2003, Mr. Mulroy was an investment banker at Citigroup and Merrill Lynch. He is also a member of the Board of Trustees of the Pegasus School, an independent primary school in Orange County, California. From January 2017 to July 2019, Mr. Mulroy served as a member of the board of directors of AgeX Therapeutics, Inc. (AGE), a biotechnology company focused on the development and commercialization of novel therapeutics targeting human aging. Mr. Mulroy earned his J.D. degree from the University of California, Los Angeles and his B.A. degree in economics from the University of Chicago. Mr. Mulroy brings to our Board his experience as the Chief Executive Officer of a publicly traded biotechnology company and member of a senior management team of a larger biopharmaceutical company that experienced a period of rapid growth. Mr. Mulroy also brings to our Board his experience in corporate finance and investor relations.
| 111 |
Angus C. Russell. Mr. Russell served as the Chief Executive Officer of Shire plc (SHPG), a biopharmaceutical company, from June 2008 to April 2013. Mr. Russell served as the Chief Financial Officer of Shire from 1999 to 2008 and also served as its Principal Accounting Officer and Executive Vice President of Global Finance. Prior to joining Shire, Mr. Russell served at ICI, Zeneca, and AstraZeneca for 19 years, most recently as Vice President of Corporate Finance at AstraZeneca plc (AZN). Mr. Russell also serves as Chairman of the Board of Directors of Mallinckrodt plc (MNK) and Revance Therapeutics, Inc. (RVNC) and as a director of Therapeutics MD, Inc. (TXMD). Mr. Russell previously served as a director of Shire plc, Questcor Pharmaceuticals, Inc. (QCOR) until it was acquired by Mallinckrodt plc (MNK) in 2014, and InterMune, Inc. (ITMN) prior to its acquisition by Roche Holdings, Inc. (RHHBY) in 2014. Mr. Russell holds an honorary Doctor of Business Administration from Coventry University, U.K. Mr. Russell brings to our Board numerous years of experience as a Chief Executive Officer of an international publicly traded specialty biopharmaceutical company and his substantial experience as an officer and director in the specialty pharmaceutical industry.
Executive Officers
Set forth below are the names, ages, offices held, tenure and certain biographical information of each of our executive officers as of March 11, 2021.
| Name | Age | Office(s) | Officer Since | |||
| Brian M. Culley | 49 | Chief Executive Officer, Interim Chief Financial Officer, and Director | September 2018 | |||
| Chase C. Leavitt | 39 | General Counsel and Corporate Secretary | May 2019 | |||
| Gary S. Hogge, D.V.M., Ph.D. | 53 | Senior Vice President of Clinical & Medical Affairs | March 2019 |
Mr. Culley’s biographical information is included above with those of the other members of our Board.
Chase C. Leavitt. Mr. Leavitt joined Lineage as General Counsel and Corporate Secretary in May 2019. Prior to joining Lineage, Mr. Leavitt served as Vice President of Legal Affairs of Tang Capital Management, LLC, a life sciences-focused investment company, and its affiliate Odonate Therapeutics, Inc. (ODT), a publicly traded biotechnology company, from June 2018 to May 2019. From May 2017 to May 2018, Mr. Leavitt served as the Deputy General Counsel of Switch, Inc. (SWCH), a publicly traded technology company, and previously served as its Associate General Counsel from July 2014 to May 2017. From 2007 to 2014, Mr. Leavitt was a corporate attorney at Latham & Watkins LLP, where his practice focused on public company representation, mergers and acquisitions and capital markets, serving life sciences and technology companies. Mr. Leavitt received a B.S. degree in business administration and a J.D. from the University of Southern California and is admitted to practice law by the State Bar of California.
Gary Hogge, D.V.M., Ph.D. Dr. Hogge joined Lineage as Senior Vice President of Clinical and Medical Affairs in February 2018. Dr. Hogge has more than 20 years of experience developing and supporting the commercialization of a number of products over a broad range of therapeutic areas. Dr. Hogge has held a variety of roles of increasing responsibility across multiple therapeutic areas in both clinical development and medical affairs. Previously Dr. Hogge was the Vice President of Medical Affairs at Questcor Pharmaceuticals, Inc. (QCOR) and before that held multiple leadership roles in both clinical development and medical affairs at Elan Pharmaceuticals including various responsibilities in the global clinical development of Tysabri® (natalizumab) in Crohn’s disease and multiple sclerosis, and for building and leading the medical affairs function. He served as medical director following the approval and launch of Tysabri. Prior to those accomplishments, he worked in clinical development for Ceplene® (histamine dihydrochloride) at Maxim Pharmaceuticals and in the immunology research and development group at Pfizer. Dr. Hogge obtained his B.S. degree and D.V.M. from Colorado State University, his M.S. and Ph.D. from the University of Wisconsin-Madison and was a visiting scientist at the Queensland Institute of Medical Research (QIMR) in Brisbane, Australia.
| 112 |
Family Relationships; Arrangements; Legal Proceedings
There are no family relationships among any of our directors and executive officers. There are no arrangements or understandings with another person under which our directors and officers was or is to be selected as a director or executive officer. Additionally, none of our directors or executive officers is involved in any legal proceeding that requires disclosure under Item 401(f) of Regulation S-K.
Code of Ethics
We have adopted a Code of Business Conduct and Ethics (“Code of Ethics”) that applies to our principal executive officers, our principal financial officer and accounting officer, our other executive officers, and our directors. The purpose of the Code of Ethics is to promote: (i) honest and ethical conduct, including the ethical handling of actual or apparent conflicts of interest between personal and professional relationships; (ii) full, fair, accurate, timely, and understandable disclosure in reports and documents that we file with or submit to the Securities and Exchange Commission (the “SEC”) and in our other public communications; (iii) compliance with applicable governmental rules and regulations; (iv) prompt internal reporting of violations of the Code of Ethics to an appropriate person or persons identified in the Code of Ethics; and (v) accountability for adherence to the Code of Ethics. A copy of our Code of Ethics has been posted on our internet website at www.lineagecell.com. We intend to disclose any future amendments to certain provisions of our Code of Ethics, and any waivers of those provisions granted to our principal executive officer, principal financial officer, principal accounting officer or controller or persons performing similar functions, by posting the information on our website within four business days following the date of the amendment or waiver.
Audit Committee and Audit Committee Financial Expert
Our Audit Committee is established in accordance with Section 3(a)(58)(A) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Our Board has determined that each member of our Audit Committee: (i) is able to read and understand fundamental financial statements, including our balance sheet, income statement and cash flow statement; and (ii) qualifies as an “audit committee financial expert,” as defined in Item 407(d)(5) of Regulation S-K; and (iii) meets the independence requirements contemplated by Rule 10-3A under the Exchange Act. Ms. Andrews’ expertise is based on her experience as Chief Financial Officer and other financial roles of STAAR Surgical Company and as a senior accountant at a major accounting firm. Mr. Russell’s expertise is based on his experience as the Chief Executive Officer and Chief Financial Officer of Shire plc, a biopharmaceutical company. Mr. Mulroy’s expertise is based on his experience as Chief Executive Officer of Asterias Biotherapeutics, Inc.
Changes in Stockholder Nomination Procedures
There have been no material changes to the procedures by which stockholders may recommend nominees to our Board since such procedures were last described in our proxy statement filed with the SEC on August 7, 2020.
| ITEM 11. | EXECUTIVE COMPENSATION |
We are a “smaller reporting company” under Item 10 of Regulation S-K promulgated under the Exchange Act and the following compensation disclosure is intended to comply with the requirements applicable to smaller reporting companies. Although the rules allow us to provide less detail about our executive compensation program, our Compensation Committee is committed to providing the information necessary to help our shareholders understand its executive compensation-related decisions. Accordingly, this section includes supplemental narratives that describe our executive compensation practices.
| 113 |
Our Compensation Committee oversees our compensation and employee benefit plans and practices, including executive compensation arrangements and incentive plans and awards of stock options and other equity-based awards under the Lineage Cell Therapeutics, Inc. 2012 Equity Incentive Plan (the “2012 Plan”). Our Compensation Committee recommends to our Board the terms and amount of executive compensation and grants of equity-based awards to executives, key employees, consultants, and independent contractors. The Chief Executive Officer may make recommendations to our Compensation Committee concerning executive compensation and performance, but our Compensation Committee makes its own determination or recommendation to our Board with respect to the amount and components of compensation, including salary, bonus, and equity awards to executive officers, generally considering factors such as company performance, individual performance, and compensation paid by peer group companies.
During 2020, our Compensation Committee engaged Marsh & McLennan (“Marsh”) to provide compensation consulting services and advice to our Compensation Committee, which included market survey information and competitive market trends in employee, executive, and director compensation programs. Marsh also made recommendations to our Compensation Committee with respect to pay mix components such as salary, bonus, and equity awards, and the target market pay percentiles in which executive compensation should fall so Lineage can be competitive in executive hiring and retention.
In reviewing each executive’s overall compensation, our Compensation Committee considers an aggregate view of base salary and bonus opportunities, equity incentive grants, and the dollar value of benefits and perquisites. These factors have been balanced against our financial position and capital resources. In making 2020 compensation decisions, our Compensation Committee reviewed market data for each named executive officer’s position, compiled from Marsh, from the following peer group companies for 2020:
| Abeona Therapeutics, Inc. | Concert Pharmaceuticals, Inc. | Inovio Pharmaceuticals, Inc. |
| Aeglea Biotherapeutics, Inc. | Curis, Inc. | Mersana Therapeutics, Inc. |
| Aravive, Inc. | Fate Therapeutics, Inc. | Neon Therapeutics, Inc. |
| Athersys, Inc. | Genocea Biosciences, Inc. | NewLink Genetics Corporation |
| Bellicum Pharmaceuticals, Inc. | Geron Corporation | Proteostasis Therapeutics, Inc. |
| Calithera Biosciences, Inc. | Harpoon Therapeutics, Inc. | Syndax Pharmaceuticals, Inc. |
| Celldex Therapeutics, Inc. | Idera Pharmaceuticals, Inc. | Synlogic, Inc. |
| Chimerix, Inc. | Infinity Pharmaceuticals, Inc. |
The 2020 peer group was recommended by Marsh and consisted of companies operating in the biopharmaceutical industry, generally with fewer than 150 employees, less than $50 million in revenue, less than $400 million in market capitalization and a lead development program in Phase 1 or 2. A limited number of companies fell outside of these parameters but were included due to their having similar areas of focus.
For 2021, our Compensation Committee engaged Anderson Pay Advisors, LLC to provide compensation consulting services and advice to our Compensation Committee. In making 2021 compensation decisions, our Compensation Committee reviewed market data for each named executive officer’s position, compiled from Anderson, from an updated peer group of companies. The following companies were added to the 2021 peer group: Apellis Pharmaceuticals, Inc., Applied Genetic Technologies Corp., Atreca, Inc., G1Therapeutics, Inc., GlycoMimetics, Inc., Spero Therapeutics, Inc., UNITY Biotechnology, Inc., and Ziopharm Oncology, Inc. The following companies were removed from the 2021 peer group: Inovio Pharmaceuticals, Inc., Mersana Therapeutics, Inc., Neon Therapeutics, Inc., NewLink Genetics Corporation, Syndax Pharmaceuticals, Inc.
Summary Compensation Table
The table below shows the compensation earned by the following, who we refer to as our named executive officers, during the fiscal years indicated: (i) our principal executive officer during the year ended December 31, 2020; and (ii) our two most highly compensated executive officers other than the principal executive officer who were serving as executive officers as of December 31, 2020.
| 114 |
Name and Principal Position(1) | Fiscal Year | Salary ($) | Bonus ($)(2) | Option Awards ($)(3) | All Other Compensation ($)(4) | Total ($) | ||||||||||||||||||
| Brian M. Culley | 2020 | $ | 551,900 | $ | 262,200 | $ | 521,563 | $ | 14,250 | $ | 1,349,913 | |||||||||||||
| Chief Executive Officer and Interim Chief Financial Officer | 2019 | 535,800 | 214,300 | — | 22,642 | 772,742 | ||||||||||||||||||
| Brandi L. Roberts | 2020 | $ | 393,200 | $ | — | $ | 260,802 | $ | 14,250 | $ | 668,252 | |||||||||||||
| Former Chief Financial Officer | 2019 | 376,137 | 169,900 | 397,948 | 3,978 | 947,963 | ||||||||||||||||||
| Chase C. Leavitt | 2020 | $ | 346,300 | $ | 131,600 | $ | 260,802 | $ | 14,250 | $ | 752,952 | |||||||||||||
| General Counsel and Corporate Secretary | 2019 | 210,146 | 102,100 | 277,310 | 9,542 | 599,098 | ||||||||||||||||||
| (1) | Mr. Culley was appointed as our Chief Executive Officer on September 17, 2018, Ms. Roberts was appointed as our Chief Financial Officer on January 7, 2019, and Mr. Leavitt was appointed as our General Counsel and Corporate Secretary on May 20, 2019. Ms. Roberts resigned from the company, and Mr. Culley was appointed as Interim Chief Financial Officer, effective January 20, 2021. The amounts reported in the table for each named executive officer represent the portion of earned compensation during the period of time such officer was in service with us. |
| (2) | The 2020 amounts represent discretionary annual bonuses as described below under “Elements of Compensation.” |
| (3) | The amounts in this column represent the grant date fair value of stock options granted to the applicable individual during the applicable year. The grant date fair value and incremental fair value of the stock options were determined in accordance with ASC Topic 718, Compensation – Stock Compensation (ASC Topic 718). See Note 12, Stock-Based Awards to our consolidated financial statements included in this Report for details as to the assumptions used to determine grant date fair value of the awards. |
| (4) | The 2020 amounts in this column represent 401(k) plan company-matching contributions. |
Narrative to Summary Compensation Table
Employment Agreements and Termination of Employment & Change in Control Arrangements
Below are descriptions of the material terms of the employment arrangements entered into with our current named executive officers.
Brian M. Culley, Chief Executive Officer and Interim Chief Financial Officer
In September 2018, we entered into an employment agreement with Mr. Culley (the “Culley Agreement”). The Culley Agreement initially provided Mr. Culley with a base salary of $530,000 annually. Mr. Culley’s salary was $551,900 in 2020 and has been increased to $580,000 for 2021. Mr. Culley is also eligible to receive an annual performance bonus of up to 50% of his base salary based upon the attainment of certain corporate and individual objectives as determined by our Board or Compensation Committee. The Culley Agreement provided Mr. Culley with reimbursement for certain travel costs to our former headquarters in Alameda, California and a monthly stipend not to exceed $3,900 for housing costs near our former headquarters, each of which ceased in August 2019.
The Culley Agreement provides that if Mr. Culley’s employment is terminated without cause or he resigns for good reason, he may be eligible for certain severance payments, including the payment of an amount equal to 12 months of his base salary, his full annual bonus amount and the payment of 6 months of health insurance premiums pursuant to our group health insurance plans as provided pursuant to COBRA. If Mr. Culley’s employment is terminated without cause or he resigns for good reason within 12 months following a change of control, then he is entitled to the acceleration of all outstanding equity awards.
| 115 |
Chase C. Leavitt, General Counsel and Corporate Secretary
In May 2019, we entered into an employment agreement with Mr. Leavitt (the “Leavitt Agreement”). The Leavitt Agreement initially provided Mr. Leavitt with a base salary of $340,000 annually and a one-time sign-on bonus of $35,000. Mr. Leavitt’s salary was $346,300 for 2020 and has been increased to $356,700 for 2021. Mr. Leavitt is also eligible to receive an annual performance bonus of up to 40% of his base salary based upon the attainment of certain corporate and individual objectives as determined by our Board or Compensation Committee.
The Leavitt Agreement provides that if Mr. Leavitt’s employment is terminated without cause or he resigns for good reason, he may be eligible for certain severance payments, including the payment of an amount equal to three months of his base salary (if terminated on or before May 20, 2020) or nine months base salary (if terminated after May 20, 2020), his prorated annual bonus amount and the payment of 6 months of health insurance premiums pursuant to our group health insurance plans as provided pursuant to COBRA. If Mr. Leavitt’s employment is terminated without cause or he resigns for good reason within 12 months following a change of control, then he is entitled to the acceleration of 50% of all outstanding equity awards (if terminated on or before May 20, 2020) or all outstanding equity awards (if terminated after May 20, 2020).
Brandi L. Roberts, Former Chief Financial Officer
In January 2019, we entered into an employment agreement with Ms. Roberts (the “Roberts Agreement”). The Roberts Agreement initially provided Ms. Roberts with a base salary of $381,924 annually, which was raised by 3% to $393,200 for 2020. Ms. Roberts was also eligible to receive an annual performance bonus of up to 40% of her base salary based upon the attainment of certain corporate and individual objectives as determined by our Board or Compensation Committee.
The Roberts Agreement provides that if Ms. Roberts’ employment is terminated without cause or she resigns for good reason, she may be eligible for certain severance payments, including the payment of an amount equal to three months of her base salary (if terminated on or before January 7, 2020) or nine months base salary (if terminated after January 7, 2020), her prorated annual bonus amount, and the payment of 6 months of health insurance premiums pursuant to our group health insurance plans as provided pursuant to COBRA. If Ms. Robert’s employment is terminated without cause or she resigns for good reason within 12 months following a change of control, then she is entitled to the acceleration of 50% of all outstanding equity awards (if terminated on or before January 7, 2020) or all outstanding equity awards (if terminated after January 7, 2020).
In connection with her resignation on January 20, 2021, we entered into a Separation and Consulting Agreement with Ms. Roberts (the “Roberts Consulting Agreement”) pursuant to which Ms. Roberts agreed to assist us with finance and accounting matters and such other matters as we and Ms. Roberts may agree to from time to time. Ms. Roberts will be compensated at the rate of $250 per hour for her services under the Roberts Consulting Agreement, which fees shall not exceed $50,000 without our approval. As additional compensation for her services under the Roberts Consulting Agreement, the equity awards held by Ms. Roberts as of January 20, 2021 will continue to vest during the term of the Roberts Consulting Agreement.
Elements of Compensation
Base Salary
Our Compensation Committee or Board reviews the base salaries of our executive officers, including our named executive officers, from time to time and makes adjustments as it determines to be reasonable and necessary to reflect the scope of an executive officer’s performance, contributions, responsibilities, experience, prior salary level, position (in the case of a promotion), and market conditions.
Annual Performance Bonuses
Each of our named executive officers are eligible to receive an annual performance bonus based on a specific target bonus amount, expressed as a percentage of base salary, and our overall achievement of specific corporate goals and objectives set by our Board and Compensation Committee each year. After the end of the year, our Board and Compensation Committee conducts an annual performance review process that evaluates achievement of overall corporate goals and achievement of specific goals and objectives by each individual executive.
| 116 |
Any final bonus payments to our named executive officers are recommended by our Compensation Committee and approved by our Board (excluding Mr. Culley), which retains full discretion to adjust individual target bonus awards. The actual bonuses, if any, awarded each year may vary from target, depending on individual performance and the achievement of corporate objectives and may also vary based on other factors at the discretion of our Compensation Committee.
For 2020, the corporate performance objective categories and respective weightings toward overall corporate bonus achievement were as follows:
| (1) | advancement of product candidates (50% weighting); | |
| (2) | Investor engagement activities and total shareholder return (“TSR”) (30% weighting); | |
| (3) | business development and licensing activities (10% weighting); and | |
| (4) | capital raising activities and efficient capital deployment (10% weighting). |
In February 2021, our Board and Compensation Committee assessed each of the corporate performance objectives and determined that the company had an overall corporate achievement level of 95% for 2020. Specifically, our Board and Compensation committee considered in its assessment that the company:
| (1) | completed enrollment in its Phase 1/2a clinical study of OpRegen with encouraging preliminary signs of tolerability and efficacy; | |
| (2) | made manufacturing improvements to its OPC1 program; | |
| (3) | negotiated and executed the early exercise of its option with Cancer Research UK to bring the VAC immune-oncology program in house; | |
| (4) | outperformed its peers and the broad indices in total shareholder return during the year ended December 31, 2020, with the strong shareholder return being sustained in early 2021; and | |
| (5) | efficiently raised capital and achieved significant budget reductions. |
Our Board and Compensation Committee determined that it was appropriate to pay each of Mr. Culley and Mr. Leavitt performance bonuses for 2020 equivalent to their target amounts multiplied by the 95% corporate achievement level. Accordingly, our Board and Compensation Committee approved cash bonuses for Mr. Culley and Mr. Leavitt in the amount of $262,200 and $131,600, respectively, which will be payable in March 2021. Ms. Roberts did not receive a performance cash bonus for 2020 because she ceased serving as an executive officer.
Other Benefits
We maintain a 401(k) defined contribution employee retirement plan for all of our employees. Employee contributions are voluntary and are determined on an individual basis, limited to the maximum amounts allowable under U.S. federal tax regulations. We provide a safe harbor contribution of up to 5.0% of the employee’s compensation, not to exceed eligible limits, and subject to employee participation.
We do not have any annuity, pension or deferred compensation plan or other arrangements for our executive officers or any employees.
| 117 |
Outstanding Equity Awards at Fiscal Year-End
The following table sets forth information concerning equity awards held by our named executive officers that were outstanding as of December 31, 2020:
| Option Awards | Stock Awards | |||||||||||||||||||||||||
| Name | Grant Date | Number of securities underlying unexercised options exercisable (#) | Number of securities underlying unexercised options unexercisable (#)(1) | Option exercise price ($) | Option expiration date | Number of shares or units of stock that have not vested (#) | Market value of shares of units of stock that have not vested ($)(2) | |||||||||||||||||||
| Brian M. Culley | 9/17/2018 | 1,042,875 | 811,125 | 1.87 | 9/16/2028 | — | — | |||||||||||||||||||
| 9/17/2018 | — | — | — | — | 92,700 | 163,152 | ||||||||||||||||||||
| 3/17/2020 | — | 1,233,300 | (3) | 0.6919 | 3/17/2030 | — | — | |||||||||||||||||||
| Brandi L. Roberts | 1/7/2019 | 239,586 | 260,414 | 1.08 | 1/6/2029 | — | — | |||||||||||||||||||
| 6/30/2019 | 71,875 | 78,125 | (4) | 1.10 | 6/29/2029 | — | — | |||||||||||||||||||
| 3/17/2020 | — | 616,700 | 0.6919 | 3/17/2030 | — | — | ||||||||||||||||||||
| Chase C. Leavitt | 5/20/2019 | 118,750 | 181,250 | 1.13 | 5/19/2029 | — | — | |||||||||||||||||||
| 7/31/2019 | 49,479 | 75,521 | (5) | 1.10 | 7/31/2029 | — | — | |||||||||||||||||||
| 3/17/2020 | — | 616,700 | 0.6919 | 3/17/2030 | — | — | ||||||||||||||||||||
| (1) | Except as otherwise provided, 25% of the options vest on the first anniversary of the grant date, and the balance vest in equal monthly installments over the three years thereafter, subject to the executive’s continued services. | |
| (2) | The dollar amounts shown in this column are calculated by multiplying the number of shares shown in the adjacent column by the closing market price of our common shares as reported on NYSE American on December 31, 2020 ($1.76), the last trading day of our fiscal year. | |
| (3) | This grant was approved by the independent members of our Board in reliance on the employment inducement exemption to shareholder approval provided under the NYSE American Company Guide. | |
| (4) | 25% of the options vested on January 7, 2020, and the balance vest in equal monthly installments over the three years thereafter, subject to Ms. Roberts’ continued services. | |
| (5) | 25% of the options vested on May 20, 2020, and the balance vest in equal monthly installments over the three years thereafter, subject to Mr. Leavitt’s continued services. |
Consideration of Shareholder Advisory Vote on Executive Compensation
The results of the advisory vote of our shareholders on the compensation of our named executive officers (commonly called the “say-on-pay” vote) at our 2020 Annual Meeting of Shareholders showed that more than 94% of our shareholders that voted approved the compensation of our named executive officers during 2019. Our Compensation Committee carefully evaluated and considered the results of this advisory vote. Aligned with the voting feedback of more than two-thirds of the shares voted, our Compensation Committee concluded that our shareholders generally supported our executive pay program and we did not make significant changes to our program for 2020. Our Compensation Committee expects to continue to consider the outcome of our “say on pay” votes and our stockholders’ views when making future compensation decisions for our named executive officers.
Director Compensation
We compensate our non-employee directors for their service on our Board and on its committees with the compensation provided below. In addition, all of our non-employee directors are entitled to reimbursements for their out-of-pocket expenses incurred in attending our Board and committee meetings.
The following table shows the annual cash fees paid to the Chairman of our Board, our directors other than the Chairman, and to the directors who served on the standing committees of our Board during 2020.
| Fees Paid | ||||
| Chairman of the Board | $ | 75,000 | ||
| Director other than Chair | $ | 40,000 | ||
| Audit Committee Chair | $ | 20,000 | ||
| Audit Committee Member other than Chair | $ | 10,000 | ||
| Compensation Committee Chair | $ | 15,000 | ||
| Compensation Committee Member other than Chair | $ | 7,500 | ||
| Nominating and Corporate Governance Committee Chair | $ | 12,000 | ||
| Nominating and Corporate Governance Committee Member other than Chair | $ | 6,000 | ||
| Financial Strategy Committee Chair | $ | 160,000 | ||
| Financial Strategy Committee Member other than Chair | $ | - | ||
| 118 |
In connection with the increase of the size of our Nominating and Corporate Governance Committee from three to four members on August 4, 2020, annual cash fees for the Chair and members other than the Chair were reduced from $15,000 and $7,500, respectively to keep overall fees for such Committee consistent. The annual cash fees are paid in four equal quarterly installments, based on the director’s continued service through the last day of the applicable quarter, other than the annual cash fees paid to the Financial Strategy Committee Chair, which are paid monthly in arrears.
In addition to cash fees, our Chairman receives an annual stock option grant to purchase 70,000 common shares and all other non-employee directors receive an annual stock option grant to purchase 40,000 common shares. All grants are made under the 2012 Plan. The options vest and become exercisable one year after the grant date.
2020 Director Compensation
The following table summarizes certain information concerning the compensation paid during our fiscal year ended December 31, 2020 to each person who served as a director during that time and who was not our employee on the date the compensation was earned.
| Name | Fees Earned or Paid in Cash | Option Award(1) | Total | |||||||||
| Deborah Andrews | $ | 69,946 | $ | 19,932 | $ | 89,878 | ||||||
| Don M. Bailey | $ | 59,855 | $ | 19,932 | $ | 79,787 | ||||||
| Neal C. Bradsher | $ | 50,185 | $ | 19,932 | $ | 70,117 | ||||||
| Stephen C. Farrell(2) | $ | 38,906 | $ | 19,932 | $ | 58,838 | ||||||
| Alfred D. Kingsley | $ | 235,000 | $ | 34,881 | $ | 269,881 | ||||||
| Michael H. Mulroy | $ | 58,071 | $ | 19,932 | $ | 78,003 | ||||||
| Angus C. Russell | $ | 52,058 | $ | 19,932 | $ | 71,990 | ||||||
| (1) | The dollar amounts in this column represent the aggregate fair market value of such awards determined based on the price of our common shares on the grant date in accordance with ASC Topic 718, Compensation-Stock Compensation (ASC Topic 718). See Note 12 Stock-Based Awards to our consolidated financial statements included in our Form 10-K for details as to the assumptions used to determine the fair value of the awards. As of December 31, 2020, the aggregate number of option awards outstanding for Ms. Andrews and Messrs. Bailey, Bradsher, Farrell, Kingsley, Mulroy, and Russell was 178,880, 140,000, 178,880, 0, 350,120, 178,880 and 178,880, respectively. | |
| (2) | Mr. Farrell did not stand for re-election and was no longer a director as of September 22, 2020. |
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT, AND RELATED STOCKHOLDER MATTERS |
The tables below sets forth certain information, as of March 5, 2021, regarding the beneficial ownership of our common shares for: (i) each person known by us to be the beneficial owner of more than 5% of our common stock; (ii) each of our directors; (iii) each of our named executive officers; and (iv) all of our current directors and executive officers as a group.
| 119 |
We have determined beneficial ownership in accordance with applicable SEC rules, and the information reflected in the table below is not necessarily indicative of beneficial ownership for any other purpose. Under applicable SEC rules, beneficial ownership includes any shares of common stock as to which a person has sole or shared voting power or investment power and any shares of common stock which the person has the right to acquire within 60 days after the date set forth in the paragraph above through the exercise of any option, warrant or right or through the conversion of any convertible security. Unless otherwise indicated in the footnotes to the table below and subject to community property laws where applicable, we believe, based on the information furnished to us and on SEC filings, that each of the persons named in table below has sole voting and investment power with respect to the shares indicated as beneficially owned.
The information set forth in the tables below is based on 161,637,890 common shares issued and outstanding on March 5, 2021. In computing the number of common shares beneficially owned by a person and the percentage ownership of that person, we deemed to be outstanding all common shares subject to options, warrants, rights or other convertible securities held by that person that are currently exercisable or will be exercisable within 60 days after such date. We did not deem these shares outstanding, however, for the purpose of computing the percentage ownership of any other person. Except as otherwise noted, the address for each person listed in the table below is c/o Lineage Cell Therapeutics, Inc., 2173 Salk Avenue, Suite 200, Carlsbad, CA 92008.
| Name and Address of Beneficial Owner | Number of Shares Beneficially Owned | Percentage of Shares Beneficially Owned | ||||||
| Greater than 5% Holders | ||||||||
| Broadwood Partners, L.P.(1) | 34,207,167 | 21.1 | % | |||||
| Named Executive Officers and Directors | ||||||||
| Neal C. Bradsher(1) | 34,207,167 | 21.1 | % | |||||
| Alfred D. Kingsley(2) | 7,107,262 | 4.4 | % | |||||
| Brian M. Culley(3) | 1,732,445 | 1.1 | % | |||||
| Brandi L. Roberts(4) | 537,651 | * | ||||||
| Chase C. Leavitt(5) | 375,667 | * | ||||||
| Michael H. Mulroy(6) | 365,715 | * | ||||||
| Angus C. Russell(7) | 206,380 | * | ||||||
| Don M. Bailey(8) | 193,970 | * | ||||||
| Deborah Andrews(9) | 148,880 | * | ||||||
| All executive officers and directors as a group (10 persons)(10) | 45,280,465 | 27.4 | % | |||||
* Less than 1%
| (1) | Includes: (i) 34,005,379 shares owned by Broadwood Partners, L.P.; (ii) 62,908 shares owned by Neal C. Bradsher; and (iii) 138,880 shares that may be acquired by Mr. Bradsher upon the exercise of options that are presently exercisable or may become exercisable within 60 days of March 5, 2021. Broadwood Capital, Inc. is the general partner of Broadwood Partners, L.P., and Mr. Bradsher is the President of Broadwood Capital, Inc. Mr. Bradsher and Broadwood Capital, Inc. may be deemed to beneficially own the shares that Broadwood Partners, L.P. owns. Mr. Bradsher disclaims beneficial ownership of the shares held by Broadwood Partners, L.P. except to the extent of his pecuniary interest therein. The Address of the foregoing entities and Mr. Bradsher is c/o Broadwood Capital, Inc., 142 West 57th Street, 11th Floor, New York, New York 10019. | |
| (2) | Includes: (i) 1,043,346 shares owned by Greenbelt Corporation; (ii) 375,351 shares owned by Greenway Partners, L.P.; (iii) 5,408,445 shares owned solely by Alfred D. Kingsley; and (iv) 280,120 shares that may be acquired by Mr. Kingsley upon the exercise of options that are presently exercisable or may become exercisable within 60 days of March 5, 2021. Mr. Kingsley controls Greenbelt Corp. and Greenway Partners, L.P. and may be deemed to beneficially own the shares that Greenbelt Corp. and Greenway Partners, L.P. own. | |
| (3) | Includes: (i) 185,602 shares held directly by Mr. Culley; (ii) 1,531,393 shares that may be acquired upon the exercise of options that are presently exercisable or that may become exercisable within 60 days of March 5, 2021; and (iii) 15,450 shares underlying RSU awards that are scheduled to vest on March 31, 2021. Does not include 77,250 shares underlying RSU awards that are subject to vesting more than 60 days after March 5, 2021. | |
| (4) | Includes: (i) 5,000 shares held directly by Ms. Roberts; and (ii) 532,651 shares that may be acquired upon the exercise of options that are presently exercisable or that may become exercisable within 60 days of March 5, 2021. |
| 120 |
| (5) | Includes: (i) 5,000 shares held directly by Mr. Leavitt; and (ii) 370,667 shares that may be acquired upon the exercise of options that are presently exercisable or that may become exercisable within 60 days of March 5, 2021. | |
| (6) | Includes: (i) 226,835 shares held directly by Mr. Mulroy; and (ii) 138,880 shares that may be acquired upon the exercise of options that are presently exercisable or that may become exercisable within 60 days of March 5, 2021. | |
| (7) | Includes: (i) 67,500 shares held directly by Mr. Russell; and (ii) 138,880 shares that may be acquired upon the exercise of options that are presently exercisable or that may become exercisable within 60 days of March 5, 2021. | |
| (8) | Includes: (i) 62,647 shares held directly by Mr. Bailey; (ii) 100,000 shares that may be acquired upon the exercise of options that are presently exercisable or that may become exercisable within 60 days of March 5, 2021; and (iii) 31,323 shares that may be acquired upon the exercise of warrants that are presently exercisable. | |
| (9) | Includes: (i) 10,000 shares held directly by Ms. Andrews; and (ii) 138,880 shares that may be acquired upon the exercise of options that are presently exercisable or that may become exercisable within 60 days of March 5, 2021. | |
| (10) | Includes: (i) 41,486,458 shares held directly by such executive officers and directors; (ii) 3,747,234 shares that may be acquired upon the exercise of options that are presently exercisable or that may become exercisable within 60 days of March 5, 2021; (iii) 31,323 shares that may be acquired upon the exercise of warrants that are presently exercisable; and (iv) 15,450 shares underlying RSU awards that are subject to vest on March 31, 2021. Does not include 77,250 shares underlying RSU awards that are subject to vesting more than 60 days after March 5, 2021. |
Equity Compensation Plan Information
The following table shows certain information concerning the options outstanding and available for issuance under all of our compensation plans and agreements as of December 31, 2020 (in thousands, except weighted average exercise prices):
| Plan Category | Number of Shares to be Issued Upon Exercise of Outstanding Options and Vesting of Restricted Stock Units, and Rights | Weighted Average Exercise Price of the Outstanding Options, and Rights | Number of Shares Remaining Available for Future Issuance under Equity Compensation Plans | |||||||||
| Equity Compensation Plans Approved by Shareholders | 14,011 | $ | 1.53 | 8,002 | ||||||||
| Equity Compensation Plans Not Approved by Shareholders(1) | 1,854 | $ | 1.87 | — | ||||||||
| Total | 15,865 | $ | 1.57 | 8,002 | ||||||||
| (1) | Reflects an option grant approved by the independent members of our Board in reliance on the employment inducement exemption to shareholder approval provided under the NYSE American Company Guide. |
The following table shows certain information concerning the options outstanding and available for issuance under all of our compensation plans and agreements for our consolidated subsidiary companies as of December 31, 2020 (in thousands, except weighted average exercise prices):
| Plan Category | Number of Shares to be Issued Upon Exercise of Outstanding Options and Vesting of Restricted Stock Units, and Rights | Weighted Average Exercise Price of the Outstanding Options, and Rights | Number of Shares Remaining Available for Future Issuance under Equity Compensation Plans | |||||||||
| Asterias Equity Compensation Plans Approved by Shareholders(1) | 350 | $ | 1.57 | 4,840 | ||||||||
| (1) | Lineage is the sole shareholder. In connection with its acquisition of Asterias, Lineage assumed sponsorship of the Asterias 2013 Equity Incentive Plan, with references to Asterias and Asterias common stock therein to be deemed references to Lineage and Lineage common shares, respectively. |
| 121 |
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
Related Person Transactions
Since July 1, 2009, Alfred D. Kingsley has made available to us the use of approximately 900 square feet of office space in New York City. We pay the office building owner $5,050 per month for the use of the space. These monthly payments are expected to cease in March 2021 when the office space lease expires.
In April 2019, Lineage issued 251,835 common shares of Lineage to Broadwood Partners, L.P., a shareholder of Lineage and Asterias Biotherapeutics, Inc. (“Asterias”), in exchange for the settlement of warrants to purchase shares of Asterias common stock in connection with our acquisition of Asterias (the “Asterias Merger”).
In connection with the putative shareholder class action lawsuits filed in February 2019 and October 2019 challenging the Asterias Merger, Lineage has agreed to pay for the legal defense of Neal Bradsher, director, and Broadwood Partners, L.P., a shareholder of Lineage, and Broadwood Capital, Inc., which manages Broadwood Partners, L.P., all of which were named in the lawsuits. During the year ending December 31, 2020, Lineage has incurred a total of $359,000 in legal expenses on behalf of the director, shareholder and the manager of the shareholder.
As
part of a financing transactions in which there were multiple other purchasers, Broadwood Partners, L.P.
purchased 1,000,000, 2,000,000, and 623,090 shares of common stock of OncoCyte Corporation (“OncoCyte”) from Lineage
in July 2019, September 2019, and January 2020, respectively.
During 2019, we invoiced OncoCyte $1.2 million for certain “Use Fees” and other charges under the terms of a Shared Facilities and Services Agreement (the “Shared Facilities Agreement”) between Lineage and OncoCyte. Under the Shared Facilities Agreement, Lineage allowed OncoCyte to use Lineage’s premises and equipment located at Alameda, California for the sole purpose of conducting business. Lineage also provided accounting, billing, bookkeeping, payroll, treasury, payment of accounts payable, and other similar administrative services to OncoCyte. The Shared Facilities Agreements also allowed Lineage to provide the services of attorneys, accountants, and other professionals who may provide professional services to Lineage. Lineage also provided OncoCyte with the services of laboratory and research personnel, including Lineage employees and contractors, for the performance of research and development work for OncoCyte at the premises. Shared services with OncoCyte were terminated with respect to the use of Lineage’s office and laboratory facilities on September 30, 2019, and December 31, 2019 with respect to all other remaining shared services.
We entered into a similar Shared Facilities Agreement with AgeX in 2018. During 2019, we invoiced AgeX $0.9 million for certain “Use Fee” and other charges and expenses for that period. Shared services with AgeX were terminated on July 31, 2019 with respect to the use of Lineage’s office and laboratory facilities and September 30, 2019 with respect to all other remaining shared services.
At the time of our acquisition of Asterias, two of our directors, Alfred D. Kingsley, and Michael H. Mulroy, and an officer of Broadwood, were directors of Asterias. Immediately following the acquisition, Don M. Bailey joined our Board, and Edward D. Wirth, III, M.D., Ph.D. joined as our Chief Medical Officer. Mr. Bailey was a director of Asterias, and Dr. Wirth was an executive officer of Asterias. All of our directors and executive officers (including Mr. Bailey and Dr. Wirth) and 5% Shareholders as reported in this report, in the aggregate beneficially owned approximately 12% of the outstanding shares of Asterias common stock as of December 31, 2018, and approximately 12% of the outstanding shares of Asterias common stock immediately prior to the acquisition on March 8, 2019.
| 122 |
Mr. Kingsley is a director of OncoCyte. Broadwood Partners, L.P., a shareholder of Lineage (“Broadwood”) beneficially owns more than 20% of the outstanding common stock of OncoCyte, and all of our directors and executive officers and 5% Shareholders as reported in this report, including Neal C. Bradsher who may be deemed to beneficially own the shares owned by Broadwood, in the aggregate beneficially own more than 20% of the outstanding shares of OncoCyte common stock. The fact that certain of our executive officers and directors own shares of OncoCyte common stock should not be considered to mean that they constitute or are acting in concert as a “group” with respect to those shares or that they otherwise share power or authority to vote or dispose of the shares that each of them own. All decisions of Lineage regarding transactions in shares of OncoCyte are made by an independent committee of our Board in which Messrs. Kingsley and Bradsher do not participate.
Related Person Transaction Policy
We have adopted a Related Person Transaction Policy that applies to transactions exceeding $120,000 in which any of our officers, directors, 5% Shareholders, or any member of their immediate family, has a direct or indirect material interest, determined in accordance with the policy (a “Related Person Transaction”). A Related Person Transaction must be reported to our Chief Financial Officer and General Counsel or outside legal counsel and will be subject to review and approval by our Audit Committee prior to effectiveness or consummation, to the extent practical. In addition, any Related Person Transaction that is ongoing in nature will be reviewed by our Audit Committee annually to ensure that the transaction has been conducted in accordance with any previous approval and that all required disclosures regarding the transaction are made.
As appropriate for the circumstances, our Audit Committee will review and consider:
| ● | the interest of the officer, director, beneficial owner of more than 5% of our common shares, or any member of their immediate family (“Related Person”) in the Related Person Transaction; | |
| ● | the approximate dollar value of the amount involved in the Related Person Transaction; | |
| ● | the approximate dollar value of the amount of the Related Person’s interest in the transaction without regard to the amount of any profit or loss; | |
| ● | whether the transaction was undertaken in the ordinary course of our business; | |
| ● | whether the transaction with the Related Person is proposed to be, or was, entered into on terms no less favorable to us than terms that could have been reached with an unrelated third party; | |
| ● | the purpose of, and the potential benefits to the transaction to us; and | |
| ● | any other information regarding the Related Person Transaction or the Related Person in the context of the proposed transaction that would be material to investors in light of the circumstances of the particular transaction. |
Our Audit Committee will review all relevant information available to it about a Related Person Transaction. Our Audit Committee may approve or ratify the Related Person Transaction only if our Audit Committee determines that, under all of the circumstances, the transaction is in, or is not in conflict with, our best interests. Our Audit Committee may, in its sole discretion, impose such conditions as it deems appropriate on us or the Related Person in connection with approval of the Related Person Transaction.
A copy of our Related Person Transaction Policy can be found on our website at www.lineagecell.com.
| ITEM 14. | PRINCIPAL ACCOUNTING FEES AND SERVICES |
The following table shows the fees billed or expected to be billed by OUM & Co. LLP (“OUM”), our principal accountant, for the audit of our annual consolidated financial statements for our last two fiscal years and for other services rendered by OUM during our last two fiscal years.
| 2020 | 2019 | |||||||
| Audit Fees(1) | $ | 319,000 | $ | 503,000 | ||||
| Audit Related Fees(2) | 50,000 | 37,000 | ||||||
| Total Fees | $ | 369,000 | $ | 540,000 | ||||
| (1) | Audit Fees consist of fees billed or expected to be billed for professional services rendered for the audit of the consolidated annual financial statements of Lineage and its several subsidiaries included in our Annual Report on Form 10-K, the reviews of the interim consolidated financial statements included in our Quarterly Reports on Form 10-Q, and services that are normally provided by our independent registered public accountants in connection with statutory and regulatory filings or engagements. | |
| (2) | Audit-Related Fees consist of fees billed for assurance and related services that are reasonably related to the performance of the audit or review of Lineage’s consolidated financial statements and are not reported under “Audit Fees.” This category includes fees related to non-routine SEC filings. |
| 123 |
Pre-Approval of Audit and Permissible Non-Audit Services
Our Audit Committee requires pre-approval of all audit and non-audit services. Other than de minimis services incidental to audit services, non-audit services shall generally be limited to tax services such as advice and planning and financial due diligence services. All fees for such non-audit services must be approved by the Audit Committee, except to the extent otherwise permitted by applicable SEC regulations. Our Audit Committee may delegate to one or more designated members of our Audit Committee the authority to grant pre-approvals, provided such approvals are presented to our Audit Committee at a subsequent meeting.
Director Independence
Our Board has determined that Deborah Andrews, Don M. Bailey, Neal C. Bradsher, Michael H. Mulroy, and Angus C. Russell qualify as “independent” in accordance with Section 803(A) of the NYSE American Company Guide. The members of our Audit Committee meet the additional independence standards under Section 803(B)(2) of the NYSE American Company Guide and Section 10A-3 under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and the members of our Compensation Committee meet the additional independence standards under Section 805(c)(1) of the NYSE American Company Guide.
Brian M. Culley does not qualify as “independent” under Section 803(A) of the NYSE American Company Guide because he is our Chief Executive Officer and Interim Chief Financial Officer. Alfred D. Kingsley does not qualify as “independent” under Section 803(A) of the NYSE American Company Guide because he was an employee of a subsidiary of ours during the past three years. Specifically, Mr. Kingsley served as Executive Chairman of AgeX Therapeutics, Inc., which was our consolidated subsidiary until August 30, 2018.
PART IV
| ITEM 15. | EXHIBITS, FINANCIAL STATEMENT SCHEDULES |
(a)(1) Financial Statements.
The following financial statements of Lineage are filed in this Report:
Consolidated Balance Sheets
Consolidated Statements of Operations
Consolidated Statements of Comprehensive Loss
Consolidated Statements of Changes in Shareholders’ Equity
Consolidated Statements of Cash Flows
Notes to Consolidated Financial Statements
(a)(2) Financial Statement Schedules
There are no financial statement schedules provided because the information called for is either not required or is shown either in the financial statements or the notes thereto.
| 124 |
(a)(3) Exhibits.
| 125 |
^ The schedules and exhibits to the merger agreement have been omitted pursuant to Item 601(b)(2) of Regulation S-K. A copy of any omitted schedule and/or exhibit will be furnished to the Securities and Exchange Commission upon request.
* Filed herewith
# Furnished herewith
+ Indicates management contract or compensatory plan
† Portions of this exhibit have been omitted pursuant to a request for confidential treatment
†† Portions of this exhibit have been omitted because the omitted information is: (i) not material; and (ii) would likely cause competitive harm to the registrant if publicly disclosed.
ITEM 16. FORM 10-K SUMMARY
None
| 126 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized on the 11th day of March 2021.
| LINEAGE CELL THERAPEUTICS, INC. | ||
| By: | /s/ Brian M. Culley | |
| Brian M. Culley | ||
| Chief Executive Officer | ||
| Signature | Title | Date | ||
| /s/ Brian M. Culley | Chief Executive Officer and Director | March 11, 2021 | ||
| BRIAN M. CULLEY | (Principal Executive and Financial Officer) | |||
| /s/ Alexandra Hernandez | Senior Director, Finance | March 11, 2021 | ||
| ALEXANDRA HERNANDEZ | (Principal Accounting Officer) | |||
| /s/ Deborah Andrews | Director | March 11, 2021 | ||
| DEBORAH ANDREWS | ||||
| /s/ Don M. Bailey | Director | March 11, 2021 | ||
| DON M. BAILEY | ||||
| /s/ Neal C. Bradsher | Director | March 11, 2021 | ||
| NEAL C. BRADSHER | ||||
| /s/ Alfred D. Kingsley | Director | March 11, 2021 | ||
| ALFRED D. KINGSLEY | ||||
| /s/ Michael H. Mulroy | Director | March 11, 2021 | ||
| MICHAEL H. MULROY | ||||
| /s/ Angus C. Russell | Director | March 11, 2021 | ||
| ANGUS C. RUSSELL |
| 127 |
Similar companies
See also AMGEN INC - Annual report 2024 (10-K 2024-12-31) Annual report 2025 (10-Q 2025-03-31)See also GILEAD SCIENCES, INC. - Annual report 2022 (10-K 2022-12-31) Annual report 2023 (10-Q 2023-09-30)
See also Moderna, Inc. - Annual report 2022 (10-K 2022-12-31) Annual report 2024 (10-Q 2024-06-30)
See also BioNTech SE
See also BIOGEN INC. - Annual report 2023 (10-K 2023-12-31) Annual report 2025 (10-Q 2025-03-31)